| Pages:
1
2
3
4 |
Hey Buddy
Hazard to Others

Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
New melt-cast secondary explosive
Firstly I wanted to apologize for previous posts I've made that were immature and distracting. I drink a little too much, the result is drinky posts.
I commit to adding more useful, less distracting input.
After some experimentation, I want to report a new secondary explosive that possesses what I believe to be beneficial qualities for amateur chemists
and excellent qualities in terms of utility, performance and production simplicity. I believe this compound is:
2-nitrimino tri-nitroso 1,3,5 triazine,
C3H4N8O5 232.1145g/mol, OB (CO2) -20%.
Assuming this to be the compound, and that I haven't really-screwed this up, I've been calling it NTNT in reference.
This explosive is a sparkling yellow crystal (somewhat whiter than TMTN and with greater light reflection by the crystals) which packs well and has an
absolute melting temp of approximately 110C. I have not measured its decomposition temperature but suspect it to be in the 220C-250C range.
Melt NTNT has a deep yellow color and reverts back to a lighter opaque lemon color at ~ 68C with a considerably increased melt-cast density.
I have not observed a significant increase in sensitivity in the melt-phase like as seen in ETN.
On exposure to flame, NTNT deflagrates without detonation in 1-2 gram quantities with intensity approximate to HMX. Deflagration leaves behind some
residue.
NTNT can be detonated with impact of a very strong hammer blow. It is less impact sensitive than PETN.
I do not believe NTNT is friction sensitive.
My analytical ability is crude, but if the 2-Nitrimino structure is correct, then NTNT has a CO2 Oxygen Balance of -20.67%.
NTNT has a Pyro Valence in its end state of (+6) for CO2 and ZERO for carbon monoxide. This is identical pyro valance for HMX and RDX which are both
zero with respect to CO, whereas Keto RDX is -6, (a net oxidizer effect depending on decomposition path) and TMTN R-Salt is (+6) for CO and (+12) for
CO2.
Assuming the structure is correct, the molar mass of NTNT would be 232.1145g/mol
NTNT is not hygroscopic, it does not solvate in H2O even after 4 days of submersion at ~15C.
NTNT is soluble in Acetone and Methanol.
It's too preliminary to declare, I suspect NTNT to have a detonation velocity and pressure higher than RDX and possibly higher than HMX or equivalent.
There is a fair chance it exceeds HMX performance. The nearest compound that I have found in literature is 2-nitriminohexahydro-1,3,5-triazine
monohydrochloride which has a detonation velocity of 9000m/sec @1.80g/cm^3.
The monohydrochloride was found in:
"Synthesis of Polynitrocompounds from Nitroguanidine" Yu Yongzhong Propellants, Explosives, Pyrotechnics 14,150-152 (1989)
Im currently testing compatibility with Al, TiH2, Cu.
Personally, I'm very excited with ease and positivity of this material's melt-castability. Even if the performance were lower than commercial
explosives, its ease of production and castability make it very useful.
It should be noted that NTNT decomposes with deflagration on even trace contact with concentrated H2SO4.
NTNT has no reaction with HCl or Glacial Acetic Acid.
It is probably carcinogenic and mutagenic.
Any help in confirmation on structure or measurements of velocity/pressure or production improvements would be welcome and I freely deliver this
information to the SM users for their enjoyment. I do have access to blast ranges for measurement and I can afford measuring equipment but I'm totally
ignorant on how to properly measure detonation velocity practically with oscilloscopes or transducers. Particularly I have somehow been unable to
measure its density with accuracy and keep getting erratic results.
The methods I have used for production are modified procedures from Lawrence Livermore synth of R-Salt. In this adaptation a slight molar excess of
Nitroguanidine to Hexamine is added to HCl and the Nitroguanidine is dissolved completely before adding additional water and the nitrite is added
after hexamine and Nitroguanidine have been mixed with water and crushed ice. The addition of NaNO2 causes the reaction and transformation from the
Nitrimino Hexahydro Monohydrochloride to the Nitrimino tri-Nitroso which results in a significant amount of light yellow thick polymeric foam to
precipitate. The foaming is greater and thicker than the foaming encountered in preparation of R-Salt. Originally, I used an overhead stirrer for this
reaction but after many trials determined it wasn't necessary and that the reaction takes place between the foam layer and the aq. HCl layer and does
not suffer in yield from using only initial stirring mixing or shaking and then leaving to react for ~25min -35m. The foam is then separated from the
liquid and dried, recrystallized from acetone.
Despite the compounds extreme decomposition sensitivity to H2SO4 (a spoonful dropped into concentrated H2SO4 results in a fireball), I found that the
highest yield and shortest reaction times can be obtained from using H2SO4 to dissolve NQ followed by dilution with H20 and addition of Hexamine to
that followed by addition of aq. NaNO2. This results in a more extreme and fast reaction and what appears to be a larger yield of product. Reaction
time with this method is only 5 minutes and the excess NaNO2 is added in stages over 5 minutes in order to prevent loss of nitrous. The foam
precipitate has to be washed thoroughly in filtration or else the NTNT will decompose upon drying with what I assume is trace H2SO4 left on the
crystals. It is also possible to recrystallize it in Acetone before it's completely dry and this eliminates H2SO4 when it is crashed out into water
with Urea as scavenger.
In conclusion, this is a low temperature (110C) melt cast explosive unknown in literature that is simple to make and can be made using either
NQ/HCl/Hexamine/NaNO2 or GN/H2SO4/Hexamine/NaNO2 with a total reaction time of 1 hr for NQ, and 10 minutes for the NTNT and those two reactions can be
performed sequentially or separated for convenience. The procedure requires no special tools or equipment. I have produced NTNT using only a large
mason jar, a pyrex baking pan and an automotive funnel with coffee filters in relatively high yield. I am still experimenting with the procedure but
it is very flexible and I'm sure others with more experience can improve on it. The resulting melt-castable NTNT produced should have a better OB
(-20.67) than both RDX (-21.6%) and HMX (-21.6%) and has a neutral pyro valance with respect to CO as neither fuel rich nor lean. This explosive may
have similar performance to RDX or even HMX, certainly much better performance than TMTN R-Salt and has reasonable sensitivities that are
less-sensitive to impact and friction than PETN.
Hopefully I have not made serious errors in reporting this information but I have no way of knowing its accuracy without review from knowledgeable
people. Any help with measurements and performance data or production experiences would be very welcome.
[Edited on 4-11-2022 by Hey Buddy]
|
|
|
boyhakan
Harmless
Posts: 2
Registered: 29-4-2019
Member Is Offline
|
|
It could be usefull to have a complete syntesis procedure so anyone, first of all me, could replicate and valuate it analitically
|
|
|
Loptr
International Hazard
Posts: 1348
Registered: 20-5-2014
Location: USA
Member Is Offline
Mood: Grateful
|
|
Care to post some details on its synthesis?
"Question everything generally thought to be obvious." - Dieter Rams
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
modified LLNL synth:
(~.14mol) 14.5+g Nitroguanidine completely dissolved in 68ml hardware store Muriatic acid
(.14 mol) 21g Hexamine added to HCl/NQ solution
2 large handfuls of crushed Ice added to solution
250ml H2O added to solution
50g NaNO2 dissolved with minimum H2O, poured into reaction vessel
vessel is shaken or stirred to mix reagents, then reaction vessel left to sit
Visible color change as Nitrous acid is formed
Foaming is produced, volume expands significantly
gas is trapped by foam layer, occasionally burping
Reaction proceeds for ~30 minutes
Reaction is then stopped, pouring foam through filter then washing with water
Multiple filters and filtrate vessels help as the foam filters slowly
Collected NTNT in foam state can either be dried as foam or squeezed out in filter and then dried compressed.
I have also used a pyrex pan and poured reaction into it at end of addition of NaNO2 and stirring, allowing foaming to occur over larger area in pan,
then scooping out foam with a spoon and then proceeding with filtration.
Recrystallize from hot acetone into dH2O with Na2CO3 and Urea as scavenger.
Dried in plate heater @50c until no moisture is evaporated
During reaction, dilution with water is critical. If no water is added there is a large loss on yield.
I am still working on the H2SO4 procedure but it is from the "Memorial Des Poudres- Vol 33" paper posted before on SM.
It uses a large excess of NaNO2 and a slight molar excess of NQ to Hexamine. In theory, one NQ mole equivalent is required for each mole of hexamine
as the nitrimino forms in the number 2 position on the triazine anything less than one equivalent will leave hexamine to become the monohydrochloride
which detonates at around 102C or so according to literature. So adding an excess of NQ is necessary to discourage monohydrochloride and ensuring the
NQ dissolves in acid is probably the safest way to ensure it is mobil for availability in the reaction.
I dont have notes on H2SO4 experiments but referencing the french TMTN paper, they call for 1200ml H20 (I think I used less, around 500ml) .8mol H2SO4
and 1.2mol NaNO2, hexamine .2 mol. I then added to the procedure greater than .2mol NQ by first dissolving it in H2SO4 and then adding the listed
reagents along with ~250grams of crushed ice. The hexamine is added quickly prior to addition of NaNO2 in H2O solution, which was added over 5 minutes
by pouring approximately 1/5 of it into reaction every minute. Precipitation of foam is immediate.
After the addition of NaNO2 is complete the reaction is swirled minimally enough to mix then left to sit for 5 minutes.
After that the reaction product is poured through coffee filter as a thick foam and then carefully squeezed of fluid in the filter to rid excess acid.
The product is then rinsed with water well, and allowed to dry before recrystallizing from hot acetone as before.
On one trial I did not wash the filtered material well and proceeded to dry it @50c, I dry on heated glass plates then scrape off dried material.
In this instance I suppose there was residual H2SO4 on the product because as I scraped the material from the glass it became powder and occasionally
as it piled onto itself began decomposing with smoke which was confusing at first until I realized it must be residual H2SO4. I have had no
decomposition sensitivities when using HCl. The material is highly sensitive to H2SO4. I tried to oxidize it once in an NH4NO3/H2SO4 slurry and it
instantly deflagrated on addition with a large fireball rising out of the beaker.
Hopefully this information helps.
All in all, it is a very simple and forgiving production regardless of route. I have not attempted the DNPT version of this but have tested the
material in acetic acid with no reaction. I do not believe this material can be oxidized into a Nitro Derivative, I believe it is rendered into an
oxidiazine derivative that is much less stable and has none of the same desirable qualities.
Below are images from lab video, it's a cheap chinese gopro so very grainy (sorry, i didnt intend for others to view it.)
The recrystallized product is on the left, you can somewhat see the sparkling quality to it. The right image is in melt phase @110c. It has no
decomposition at all in melt phase and at ~68C it reverts back to the original lighter color in a more-dense casted state
 
[Edited on 4-11-2022 by Hey Buddy]
[Edited on 4-11-2022 by Hey Buddy]
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
Intersting description and procedure.
14.5g NQ + 68ml HCl +21g Hex + 50g NaNO2. It is approx 154 g of all reagents. And yield of grams after recrystallisation from acetone?....17g for
example?
Maybe I'm old and blind, but I can't find it in the text.
[Edited on 4-11-2022 by Laboratory of Liptakov]
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
Quote: Originally posted by Laboratory of Liptakov  | Intersting description and procedure.
14.5g NQ + 68ml HCl +21g Hex + 50g NaNO2. It is approx 154 g of all reagents. And yield of grams after recrystallisation from acetone?....17g for
example?
Maybe I'm old and blind, but I can't find it in the text.
[Edited on 4-11-2022 by Laboratory of Liptakov] |
So sorry doc, I have not recorded yield data. I immediately begin testing on it everytime I prepare it. It is improved beyond both TMTN procedures,
the yield is higher for this compound, somehow additives like guanidines and ureas increase the yield. I have performed the TMTN synths so many times
it is clearly observed there is a greater yield on product when preparing this NTNT versus TMTN. I would guess more than 17g. I'm still tweaking the
H2SO4 procedure but the modified LLNL synth is a great place to start, very simple.
I did just realize I miswrote in earlier post, to clarify, NQ molar equivalent to hexamine controls monohydrochloride formation, if there is less NQ
equivalent than the remainder of hexamine, then that remainder should become TMTN upon addition of NaNO2. The molar qty of NaNO2 controls how much
monohydrochloride (2-nitriminohexahydro-1,3,5-triazine monohydrochloride) is produced. If there is not enough NaNO2, then not all of the
monohydrochloride is converted into NTNT. So a slight excess of NQ in molar ratio to Hexamine is a good measure, and an excess of NaNO2 is an
important measure to avoid monochloride in the final product because according to Yohngzung, the monohydrochloride explodes at 102C which is below the
melt cast temp of NTNT at 110C.
The picture below helps to explain sub-reactions taking place before the formation of NTNT. These compounds can be easily produced as well though they
are not new and have less stability and much less utility.
I have attached two references for polynitro compounds for NQ. These papers are where my research started and after a lot of failed experiments with
strange derivitives I came to this NTNT compound and going through testing procedures found that it is very suitable as an explosive. I really need to
determine velocity and pressure, but for the simplicity of production it is hard to beat.

Attachment: nitroguanidine poly nitro compounds Zhuang.pdf (283kB)
This file has been downloaded 293 times
Attachment: nitroguanidine derivatives.pdf (134kB)
This file has been downloaded 281 times
[Edited on 4-11-2022 by Hey Buddy]
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
So the excess of both NQ and NaNO2 over hexamine is important. Apparently, 68 ml of HCl is probably not that important. Could it be 75ml? Or just
60ml? Does it only create an acidic reaction environment?
250 ml of water is clear info, but two handfuls of ice is not. How much ice is that?
500 grams? So a total of 750ml of distilled frozen water? So is this a reaction medium that is very dilute? Will it require a glass of at least 2
liters of container volume?
Your description......(I think I used less, about 500ml)....is not accurate enough. For anyone to even bother trying to repeat. If you are interested
in your invention being viable (and being tried by someone) the process must be described with absolute precision. 2 handfuls of ice is not enough. 2
handfuls of distilled ice? How much acetone to recrystallize 10g of LLNL? What temperature? How many grams of LLNL should be dissolved in 100g of
acetone? How much urea? How much Na2CO3? If the description isn't accurate, no one will even bother trying. Everyone will make ETN and be
satisfied.....
Years of experience say this: Amateur chemists are incredibly lazy. If they don't get all the information on a golden platter, they won't even open
the door to the lab.
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
MineMan
International Hazard
Posts: 1004
Registered: 29-3-2015
Member Is Offline
Mood: No Mood
|
|
So if a little of the monochloride is formed it can explosive the whole batch during melt casting!???
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
This I don't know. It appears possible from literature, so I've warned about it. I don't know how well the monochloride forms, or if the data on it
exploding @102C is accurate. But for safety, I would assume it to be accurate. It would need to be tested by deliberately using insufficient Nitrite
in order to create monohydrochloride then testing it with controlled heating to see if detonation occurs @102C. But I have not tested this.
The way this compound was come across was in attempting to replicate the synth of the monohydrochloride. During those work ups, I found difficulty in
precipitation. I have familiarity with TMTN precipitation methods, so that immediately came to mind as a possibility because it's a triazine. The
nitrosation with NaNO2 was very effective in promoting precipitation and I moved on to basic testing of the material.
I feel its properties and simplicity of production are significant enough to warrant sharing this information even at this early stage because it
appears to be a very interesting and capable energetic material. It could potentially offer a great deal of velocity and pressure.
Monohydrochloride formation hazard, I feel is something that should be tested for, or at least warned of as a potential hazard as it seems to be a
possible pathway in preparation of this compound if insufficient Nitrite is used.
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
| Quote: Originally posted by Laboratory of Liptakov | So the excess of both NQ and NaNO2 over hexamine is important. Apparently, 68 ml of HCl is probably not that important. Could it be 75ml? Or just
60ml? Does it only create an acidic reaction environment?
250 ml of water is clear info, but two handfuls of ice is not. How much ice is that?
500 grams? So a total of 750ml of distilled frozen water? So is this a reaction medium that is very dilute? Will it require a glass of at least 2
liters of container volume?
Your description......(I think I used less, about 500ml)....is not accurate enough. For anyone to even bother trying to repeat. If you are interested
in your invention being viable (and being tried by someone) the process must be described with absolute precision. 2 handfuls of ice is not enough. 2
handfuls of distilled ice? How much acetone to recrystallize 10g of LLNL? What temperature? How many grams of LLNL should be dissolved in 100g of
acetone? How much urea? How much Na2CO3? If the description isn't accurate, no one will even bother trying. Everyone will make ETN and be
satisfied.....
Years of experience say this: Amateur chemists are incredibly lazy. If they don't get all the information on a golden platter, they won't even open
the door to the lab. |
With all due respect, I was simply reporting the compound, it was not my intent to deliver a recipe.
When I found that the material was melt castable, non hygroscopic and burned with greater intensity than RDX, I then moved on to hammer testing and
was able to detonate it with a ~one pound hammer striking the material against a steel anvil *very hard (TMTN cannot be detonated with a hammer by
hand. it is extremely insensitive to impact).
Following that, I realized there was something significantly useful here and so I analyzed the likely structure and realized its Pyro Valence
features, high nitrogen content and saw that if the nitrimino structure were correct, it would have one less oxygen than RDX. The Nitrimino function
eliminates hydrogen at the number 2 position and so the CHNO ratios become more favorable. If that were the case, it would seem to agree with the
increased reactivity over TMTN.
The Nitrimino function tends to add quite a bit of energy to molecules as an addition. For example, some nitriminotetrazoles are reported in the
10,000m/s velocity. Amino-Nitroguanidine and ANQN and ANQ Sulfate are very energetic in themselves, so adding a nitrimino function to TMTN should in
theory be much more powerful.
My hope was that someone could assist in identifying the compound or perhaps run it through theoretical software to estimate velocity and pressure. I
was not trying to deliver a recipe though.
If SM would like, I can develop a procedure for preparation but it's really very simple, it is virtually identical to TMTN procedures which are
probably about the simplest out there aside from Nitroguanidine preparation.
For now, I am simply excited to have located a new melt cast because they are less commonly found, so I wanted to share it with anyone interested. If
chemists are too lazy to prepare compounds themselves then they can wait until a procedure is finished working up. ie. Not my problem.
|
|
|
Texium
Administrator
Posts: 4580
Registered: 11-1-2014
Location: Salt Lake City
Member Is Offline
Mood: PhD candidate!
|
|
I think LL did himself a disservice by saying that a detailed procedure should be provided because “amateur chemists are lazy.” The reason that
you should write up a detailed procedure when you claim a new compound isn’t to provide a recipe for the lazy, it’s to ensure that your results
are unambiguous and reproducible. This goes to an even greater extent in professional chemistry publications. Not only is a detailed procedure
required when reporting a new compound, but full characterization data with IR, NMR, and mass spectroscopy, and sometimes melting point and TLC Rf,
too. Obviously that full characterization is not practical for an amateur to achieve, but the percent yield should definitely be reported, and it’s
nice to see at least the melting point and Rf provided. You can probably find someone here with the resources to further characterize a sample. Nobody
is giving you shit here, it looks like you’ve made an interesting discovery. This is all just constructive criticism to help you report your results
in a professional and reproducible manner.
[Edited on 11-5-2022 by Texium]
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
Oh yes, I understand. We thanks for your research. Procedure looks pretty easily. For RDX is necessary usually almost 100% HNO3.
For your substance are necessary pretty easy, available compounds. Also procedure looks easy. Final test require of course the comparative crater
against ETN. In the lead or the aluminium block. For example 1g cast or high pressed your substance. Any way, seems it interestly.
If the substance turns out to be as explosive as ETN, or even more explosive, it would be a breakthrough in energy materials research. On amateur
field.
I hope, that on picture is Nitroguanidine - NQ
As Texium said. It’s to ensure that your results are unambiguous and reproducible.

[Edited on 5-11-2022 by Laboratory of Liptakov]
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
Loptr
International Hazard
Posts: 1348
Registered: 20-5-2014
Location: USA
Member Is Offline
Mood: Grateful
|
|
Well, the reason that you also provide the procedure is because the burden is on the one posting the topic. It goes against forum etiquette for one,
but also as Texium mentioned.
Just because you had success after unintentionally altering the procedure, doesn't mean the next will will be as successful, especially with
energetics.
[Edited on 5-11-2022 by Loptr]
"Question everything generally thought to be obvious." - Dieter Rams
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
| Quote: Originally posted by Texium | I think LL did himself a disservice by saying that a detailed procedure should be provided because “amateur chemists are lazy.” The reason that
you should write up a detailed procedure when you claim a new compound isn’t to provide a recipe for the lazy, it’s to ensure that your results
are unambiguous and reproducible. This goes to an even greater extent in professional chemistry publications. Not only is a detailed procedure
required when reporting a new compound, but full characterization data with IR, NMR, and mass spectroscopy, and sometimes melting point and TLC Rf,
too. Obviously that full characterization is not practical for an amateur to achieve, but the percent yield should definitely be reported, and it’s
nice to see at least the melting point and Rf provided. You can probably find someone here with the resources to further characterize a sample. Nobody
is giving you shit here, it looks like you’ve made an interesting discovery. This is all just constructive criticism to help you report your results
in a professional and reproducible manner.
[Edited on 11-5-2022 by Texium] |
Of course, I understand, my wife is mad at me and I cant retrieve lab notes and so I was not prepared to discuss a procedure. But I could be hit by a
bus or some unfortunate circumstance and so I wanted to put out this information for other people to know it exists. I will retrieve lab notes and
reconstruct the LLNL mod synth for this compound that has been successful. The Ice added has been tried at various amounts and they all yield product,
I think the only experiment that I tried that didnt yield product was one without any cooling whatsoever and it became very hot and produced a lot of
gas, obviously, and virtually no yield. But I stated "two handfulls" of ice because that was the note in the notes that I do have for one of the LLNL
mod trials. I will pool notes and figure out where holes are and retest melt point with a thiel apparatus and determine decomp temp. reconstruct that
with accuracy and report back with some more data. FYI, m.p. for me began at 104C and then had a sweet spot at 110C where it melted very easily, with
no decomposition or off gassing at all. This is very different from TMTN, as TMTN off gasses in a very narrow window of its melt phase making it
practically not possible to melt cast with TMTN. This compound is different entirely in that and other respects.
But I understand the complaints, I estimated there may not be much interest but if there is interest I will report back with confirmations and
simplified procedure.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
My experience is that many amateur chemists try to simplify the original procedure. And that immediately on the first try. Even if the original
procedure was simplest, there will always be someone who feels smarter than the author. And he will try to simplify the process even more.
The result is a failure of the synthesis, or a tragic yield. Or some worse.
Therefore is necessary exact procedure, which bring always same results.
Louis Pasteur: One attempt, nothing attempt. 10 attempts = one attempt.
100 attempts half of truth. 1000 attepts, the whole truth.
My experience says, that 10 attempts is enough. With same results.
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
| Quote: Originally posted by Loptr | Well, the reason that you also provide the procedure is because the burden is on the one posting the topic. It goes against forum etiquette for one,
but also as Texium mentioned.
Just because you had success after unintentionally altering the procedure, doesn't mean the next will will be as successful, especially with
energetics.
[Edited on 5-11-2022 by Loptr] |
No, in preparation of this compound the results are repeatable across a wide range of cooling conditions, so long as enough Nitrite is used to convert
it from monochloride to this compound, there should be a repeat and the only difference as far as I can ascertain is the yield, depending on cooling
conditions. I have performed this several times now for both HCl and H2SO4. Cold seems to improve yield. I have been unprepared for posting a
procedure, as for now I have moved on to using H2SO4 but this presents other complications. So I understand these criticisms and will try to put
together something more comprehensive on a procedure with HCl and report back.
Thank you for your criticisms.
If anyone has theoretical modeling software, I am interested in learning theoretical performance.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
| Quote: Originally posted by Hey Buddy |
But I understand the complaints, I estimated there may not be much interest but if there is interest I will report back with confirmations and
simplified procedure. |
Interest is evidently..... .... Caution: Keep away from bus, tram, train, open
window, bridge, elevator and similarly. Do not travel anywhere, especially not to Ukraine. .... Caution: Keep away from bus, tram, train, open
window, bridge, elevator and similarly. Do not travel anywhere, especially not to Ukraine.
If your substance had an easy DDT, it would be one of the biggest breakthroughs in the history of Science Madness.....
[Edited on 5-11-2022 by Laboratory of Liptakov]
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
Loptr
International Hazard
Posts: 1348
Registered: 20-5-2014
Location: USA
Member Is Offline
Mood: Grateful
|
|
| Quote: Originally posted by Hey Buddy | | Quote: Originally posted by Loptr | Well, the reason that you also provide the procedure is because the burden is on the one posting the topic. It goes against forum etiquette for one,
but also as Texium mentioned.
Just because you had success after unintentionally altering the procedure, doesn't mean the next will will be as successful, especially with
energetics.
[Edited on 5-11-2022 by Loptr] |
No, in preparation of this compound the results are repeatable across a wide range of cooling conditions, so long as enough Nitrite is used to convert
it from monochloride to this compound, there should be a repeat and the only difference as far as I can ascertain is the yield, depending on cooling
conditions. I have performed this several times now for both HCl and H2SO4. Cold seems to improve yield. I have been unprepared for posting a
procedure, as for now I have moved on to using H2SO4 but this presents other complications. So I understand these criticisms and will try to put
together something more comprehensive on a procedure with HCl and report back.
Thank you for your criticisms.
If anyone has theoretical modeling software, I am interested in learning theoretical performance. |
Sorry, but my points still stand and it wasn't provided as criticism towards you. LL said that amateur chemists are lazy. My point was that if we go
look up a procedure, we can not be sure it's the same one or revision. I then concluded my response with it being understood that if you want to
discuss a compound you prepared, that at a minimum, you need to include the procedure that you followed because it establishes a common reference
point for others to base their work.
Now, as for the repeatability in your experience, that might be so. My experience tells me that generally the technique, reagent purity, implicit
understanding of the reaction mechanisms, and unintended modifications to procedure by multiple people in multiple locations and timezones can have an
effect on the outcomes. But what do I know?
"Question everything generally thought to be obvious." - Dieter Rams
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
I made a video essay to make more sense
https://www.bitchute.com/video/GuXsIe7K3SX2/
LL, the yield on this recorded video is only 33%, where as TMTN is 40% via LLNL R Salt procedure. There are other tweaks that increase yields but for
starting I think it's a good baseline because it is reproducible and mirrors the LLNL procedure with the exception of the NQ @ .16 mole. Sorry I guess
greater than 17g will have to come later.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
Great video, we thanks......
12,4g is better than nothing.
The test in a solid cavity and the depth of the crater in the lead block will be decisive for the attractiveness of your substance. Or perforating a
3mm thick steel sheet with 1g of your cast substance.
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
| Quote: Originally posted by Laboratory of Liptakov |
depth of the crater in the lead block will be decisive for the attractiveness of your substance. Or perforating a 3mm thick steel sheet with 1g of
your cast substance. |
What dimension of lead block?
how is cap oriented to block (side or end)?
Is the sheet steel a standard spec?
I've found some 3mm 1018-mild steel. it appears to be 3mm nominal but actual is 2.45mm. I may be able to test this week. Now I only have PETN and ETN
that could be compared. I do not have any RDX or HMX prepared. I do have quite a bit of TMTN.
Is there a SM test procedure standards thread? I have not seen it if so.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
The cavity is always perpendicular. The steel sheet is a standard sheet, or a U or D profile, 2 - 3 mm thick. 2.45 mm is also OK. In the picture, the
lead block is 40 mm high and 65 mm in diameter. Which is suitable for the test of 300 - 1000 mg ETN, PETN, RDX. Pressed 400 mg ETN (50 kg pressure in
ID 6mm) gives repeatedly the same result as shown in the picture. If 400 mg of ETN is used, the result should be the same anywhere in the world. Or
very similar. Thread about standardized testing not exist.

[Edited on 6-11-2022 by Laboratory of Liptakov]
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: Originally posted by Laboratory of Liptakov | The cavity is always perpendicular. The steel sheet is a standard sheet, or a U or D profile, 2 - 3 mm thick. 2.45 mm is also OK. In the picture, the
lead block is 40 mm high and 65 mm in diameter. Which is suitable for the test of 300 - 1000 mg ETN, PETN, RDX. Pressed 400 mg ETN (50 kg pressure in
ID 6mm) gives repeatedly the same result as shown in the picture. If 400 mg of ETN is used, the result should be the same anywhere in the world. Or
very similar. Thread about standardized testing not exist.
[Edited on 6-11-2022 by Laboratory of Liptakov] |
What about melt-cast ETN topped with (dextrinated)NHN? I hope that is good enough!
|
|
|
Hey Buddy
Hazard to Others
Posts: 429
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
Just recorded alternate procedure for the preparation of this compound. I will add yield record to this post and add a video link later. This method
is based off of the "Memorial Des Poudres" TMTN procedure translation. I prefer it over LLNL procedure and believe it's capable of higher yield. I
will play with dilution for yield but as a baseline this is minimal.
NQ 21.85 g (0.21 mol)
Hexamine 28.03 g (0.2 mol)
NaNO2 68.99 g (1 mol)
H2SO4 53.94 g (.55 mol) (Desolvo Industrial Drain Cleaner 98%+?)
Ice (d) 350 g
H2O (d) 200 ml
21.85 g NQ is added to solution of 53.94 g H2SO4 + 100 ml H2O.
100 g Ice is added to this NQ/H2SO4 solution
NQ is dissolved into solution with stirring
28.03 g Hexamine is added to the solution
250 g of Ice is added to the solution
A second solution 68.99 g NaNO2 and 100 ml H2O
This second solution (NaNO2 + H2O) is added ~20ml per minute and is completed in 5 minutes, with five sets of addition, each one minute apart,
totaling 5 minutes.
After final addition of Nitrite, allow reaction to stand 5 minutes
Filter and rinse reaction.
The product from this method is a polymeric consistency and can hold a lot of reaction fluid in its mass. Buchner with vacuum or hand wringing filters
is necessary.
Product is recrystallized from Acetone.
Still testing metal compatibility and DDT capability, so far DDT seems unlikely. The casting ability and ease of preparation are the attraction to
this compound.
/* Nov 8 2022
Returned this morning to find partially decomposed product free of water after drying @70C overnight.
Product from H2SO4 method (with 200 ml H2O + 350 g ice) decomposes spontaneously when dry.
From prior trials it was found that damp product can be recrystallized while wet in Acetone with no issues of decomposition and minimal loss in
solvent.
It is probably possible to water bathe after initial filtration, then dry. This possibility should be considered.
Spray washing is not enough to prevent decomposition, material holds acidic liquid that is surface borne on material which then contacts itself on dry
processing and decomposes turning black and smoking.
On review of footage of this procedure, too much Red/Brown NO2 is liberated with this ratio of dilution.
350 g Ice seems to be appropriate amount to give fully melted Ice on filtering.
Temperature threshold seems wide for conditions, any temperature <20C seems to have no effect on yield.
Next step in development of this procedure is to minimize NO2 liberation, this will be tried by increasing dilution ratio with water.
Results from that will determine if dilution ratio has effect on dry decomposition or if product requires water bath and second filtering operation
prior to drying.
After that is determined and incidental H2SO4 decomp is prevented, accurate yields can be recorded. It seems certain that there is a lot more product
when using H2SO4. If TMTN methods are analogous, proper H2SO4 should raise yield to 44-50% range. */
/*NOV 8 2022
Increasing water from 200 ml to 450 ml directly reduced NO2 bleed.
There is still burping but the addition of more water tames the off gassing well. Yield also appeared to increase.
Not sure if the additional dilution alone would prevent decomposition on drying.
Opted to avoid potential acidic decomposition and bathed the filtered material in water and re-filtered. Loss appeared minimal.
Assuming H2SO4 method can be tailored for high yield despite its complications, it is preferred to HCl method if H2SO4 is available.
Reaction time is minimized to 5 minutes and yield appears increased.
Other benefits are the minimizing of reagents and potential combination of Nitroguanidine preparation with this method in a continuous process which
would be about one hour fifteen minutes in total and could avoid ever precipitating nitroguanidine from H2SO4.
Downsides are high reactivity of product with residual H2SO4, which could lead to fireball (or detonation) decomposition. Other potential
complications are the decomposition of hexamine by H2SO4 which is eliminated or limited by adding hexamine immediately prior to NaNO2 with minimal
time in solution.
Acidic liquid trapped in material during filtration could probably be helped by the use of vacuum with buchner assembly, but for now using automotive
funnel/mesh sieve/ coffee filter system which is very cheap and replicated at any location with grocery store. General process for filtration of
liquid entrapped polymeric product is: pour into coffee filter, allow gravity filtration until filtration becomes slowed to drip, then carefully
collecting the edges of paper coffee filter and bringing them together to be pinched off, preventing escape of material from the top, then gently
squeezing or "milking" bloated filter until it compresses and frees itself of entrapped liquid.
All of this will be covered in follow up video on this H2SO4 tentative procedure. It will include less successful attempt with decomp as example. Will
continue preparation trials and collecting yield data concurrently building surplus for additional testing including explosive performance.
LL, sorry but it appears to not be a good candidate for DDT so far. The primary appeal of this would be:
Insensitive Melt cast>Potential of RDX+ Performance> Relative ease of preparation. */
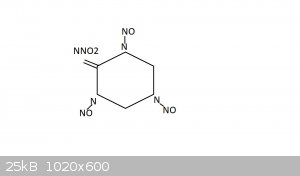
I assume structure is this, but have no idea. I drew a picture earlier but had forgotten double bonds of nitrosamine
Attachment: TMTN_Memorial_des_poudres.pdf (87kB)
This file has been downloaded 272 times
[Edited on 8-11-2022 by Hey Buddy]
[Edited on 8-11-2022 by Hey Buddy]
[Edited on 8-11-2022 by Hey Buddy]
[Edited on 8-11-2022 by Hey Buddy]

[Edited on 9-11-2022 by Hey Buddy]
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1386
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
I estimate , that H2SO4 53.94g is counted as 100% H2SO4....?....It is approx 31% concentration of H2SO4 after diluting in 100g dH2O...?...
Together you use 172.81 g of all reagents. Yields for ETN are usually 11 - 12% from all reagents.
Your method should by provide similar yield. Thus 19g - 21g TMTN.
The great advantage of your method is the use of dilute H2SO4 and the absence of HNO3. Nitroguanidine can be a disadvantage. (availability -
production)
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
| Pages:
1
2
3
4 |









