Knowledge Seeker
Harmless

Posts: 16
Registered: 15-12-2020
Member Is Offline
|
|
Benzil dioxime
Hello science madness comunity
I attempted to prepare benzil dioxime following the procedure from this paper https://doi.org/10.1139/v75-043
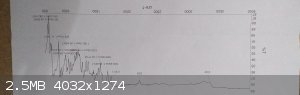
I obtained a good amount white precipitate. I initially measured its m.p and it was 241-245 c°. Then I filtered it off, washed with methanol several
times and then with hot water. When the precipitate dried I measured its melting point again and it was 250-251C°. But the IR spectrum of the
precipitate was not satisfying. Do you have any thoughts? Can anyone provide me with a method to successfully prepare benzil dioxime?
Thank you

[Edited on 28-5-2021 by Knowledge Seeker]
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Does benzil dioxime have any advantage than more common dimethylglyoxime?
Here they claim 3 forms (alfa beta gamma) with various m.p.
https://www.drugfuture.com/chemdata/benzil-dioxime.html
| Quote: | 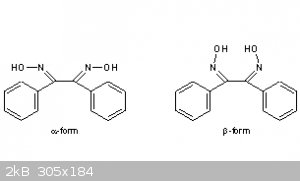
Literature References: Three isomers occur: a or anti, b or syn, g or amphi. Prepn of a-form from benzil and hydroxylamine hydrochloride: Brady,
Perry, J. Chem. Soc. 127, 2874 (1925); F. J. Welcher, Organic Analytical Reagents vol. III (Van Nostrand, New York, 1947) pp 224-227; Boyer et al., J.
Am. Chem. Soc. 77, 5688 (1955). Prepn of b-form: Brady, Perry, loc. cit.; Boyer et al., loc. cit. Prepn of g-form: Welcher, loc. cit., pp 227-228; see
also Boyer et al., loc. cit.
Derivative Type: a-Form
Properties: Crystals from methanol, mp 238-240°, Boyer et al., loc. cit., also reported as mp 243-244°, Meisenheimer, Lamparter, Ber. 57, 276
(1924). Practically insol in water, ether, glacial acetic acid; slightly sol in alc; readily sol in NaOH solns.
Melting point: mp 238-240°; mp 243-244°, Meisenheimer, Lamparter, Ber. 57, 276 (1924)
Derivative Type: b-Form
Properties: Crystals, mp 212-214°, Meisenheimer, Lamparter, loc. cit.
Melting point: mp 212-214°, Meisenheimer, Lamparter, loc. cit |
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Online
Mood: No Mood
|
|
Many mono-oximes can exist in two forms if the ketone is asymmetrical. So I guess with two adjacent oximes there are three possible positional
isomers. Apart from this there is a tendency for vicinal di-ones to form a monoxime rapidly but the dioxime more slowly so maybe you have a mixture of
di and mono oximes.
|
|
|
kmno4
International Hazard
Posts: 1504
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
Then measured m.p. would be far below 200 C and not so sharp.
Looking at IR spectrum and m.p. determination, I have impression that it could be impure oxanilide. Treating prepared compound with NaOH would confirm
(or rule out) this assumption.
According to Diehl*, it does.
*see references from the paper cited above
[Edited on 2-6-2021 by kmno4]
Слава Україні !
Героям слава !
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Online
Mood: No Mood
|
|
Hi Knowledge_seeker, kmno4’s suggestion that the main product may be oxanilide suggests that a Beckmann’s type rearrangement of the dioxime is
occurring. This possibility had not occurred to me before kmno4 suggested it but on reflection it seems like a good idea so I did a little digging and
found a review of the Beckmann rearrangement in Organic Reactions, vol. 11, p1 (attached below). It appears that the reactions of benzyl monooxime and
dioxime are complex (see pages 35 to 39) and I suggest you give it a read.
In summary the double Beckmann’s rearrangement of the dioxime to oxanilide suggested by kmno4 has not been reported though that doesn’t mean it
can’t happen! Amongst the previously reported product are things like diphenylfurazan, diphenyl-1,2,4-oxodiazole, benzamide and benzonitrile.
Since you seemed to have followed the preparation in the original paper fairly faithfully you might reasonably expect to obtain the desired product,
however, on reflection two points came to mind:
1) The paper only characterises the product by melting point while you have used IR and Mp. Their Mp may have been just fortuitous.
2) If I understand their method correctly they are measuring nickel by co-precipitation of the chelate with excess insoluble dioxime and measuring the
intensity of some specific wavelength of reflected light. So provided the insoluble co-precipitated material is white it doesn’t matter too much
what it is as along as it co-precipitates. So the fact that the compound forms a nickel complex tells us nothing about the absolute purity of their
product. Since they use a large excess the only requirements are that the insoluble portion is white and co-precipitates.
It is worth bearing in mind that generally a base is used when preparing oximes with hydroxylammonium chloride to consume the free acid liberated by
the reaction. The reaction is an equilibrium reaction so the base helps drive the reaction to completion by consuming the liberated acid. However, in
the original paper no base is used and so the free acid may accumulate and cause various side reaction including rearrangement and/or cleavage of the
oximes.
It might be worth running the reaction with the slow addition of sodium acetate, sodium carbonate or pyridine etc. during the the reaction period.
Attachment: Organic Reactions Vol 11 ch 1 Beckmann's Rearrangement Donaruma.pdf (6.2MB)
This file has been downloaded 462 times
|
|
|
kmno4
International Hazard
Posts: 1504
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
Hi Boffis. My suggestion is based only on m.p. determination.
But if Knowledge Seeker's determinations of m.p. are incorrect, giving values higher than in reality, with error about + 5 C, then it changes
everything.
IR spectra for the dioxime (and also for oxoanilide) are available from internet, but differs markedly from the pictures above.
I have impression that KS's IR masurement is not done correctly 
Nevermind.
I have some benzil, prepared long time ago. I am going to try this preparation if I have time. As Boffis noticed, orginal procedure utilizes
hydroxylamine hydrochloride alone, who knows why....
Besides the yield of the dioxime is very low, comparing to input diketone. The author couldn't afford sodium acetate, the reaction needs higly acidic
medium, or what ?
Fortunately, all substrates (but time) I have at hand 
Слава Україні !
Героям слава !
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
Quote: Originally posted by kmno4  | | As Boffis noticed, orginal procedure utilizes hydroxylamine hydrochloride alone, who knows why.... |
It goes back to 1888, for the alpha, so it's made without base at least 3 times in the lit, (only know the OAR ref, read no others mentioned here)
don't know if any mention yield...Organic Analytical Reagents (from archive.org IIRC) also has the gamma prep from someone; base is used there, so
maybe it depends on what you want.
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Online
Mood: No Mood
|
|
While searching for other information I stumbled across this short article that gives a procedure for the conversion of a complex benzil derivative to
the corresponding dioxime. In this procedure dry potassium carbonate was used to liberate free hydroxylamine. I thought it might be of interest to
someone.
Attachment: IJCB 38B(10) 1208-1210.pdf (597kB)
This file has been downloaded 361 times
|
|
|
kmno4
International Hazard
Posts: 1504
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
After making several experiments, I have to confirm low yield of a-dioxime prepared. But literature research gave me explanation of this phenomenon.
Melting poins given at the beginning of the discussion are surely incorrect (measurement error...?).
Something to read in attachment.
Reference no.119 gives 3,4 g of dioxime from 5,0 g of benzil.
The most possibly it is a typo in orginal paper (available online).
Repeating procedure from this paper gave yield very close to the yield given in quoted earlier canadian paper.
Attachment: Annotated bibliography of a-benzildioxime.pdf (1.4MB)
This file has been downloaded 363 times
Слава Україні !
Героям слава !
|
|
|
kmno4
International Hazard
Posts: 1504
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
A small addendum.
a-benzildioxime prepared on hot from benzil melts at 238-241 C.
Previous measurement gave 242-244 C but rate of heating was too large and this m.p. was slightly incorrect.
A sample of this compound was also prepared in a slightly different way (benzil -> a-benzilmonoxime -> a-benzildioxime). The yield was much
better: 1,9 g from 2,0 g of benzil, but it melts at around 230 C. However, I think it is not bad. Preparation of the monoxime can be found in Vogel
(it is rewritten, without any changes, from JCS article from 1930 y). Conversion of the monoxime into dioxime can be found in a publication from
Berichte (in attachment). In fact, separation of the (alpha) monoxime is not necessary for this purpose.
Attachment: uc1-b3481783-597-1632563808.pdf (181kB)
This file has been downloaded 271 times
Слава Україні !
Героям слава !
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
reactants:
benzil 2,10 g (10 mmol, Molar mass 210.232 g/mol)
hydoxylamine hydrochloride 1,80 g (26 mmol, 1,3 fold excess, Molar mass 69.49 g/mol)
7,5 g methanol (should be 9,5 ml, I almost always weigh liquids instead measuring their volume)
procedure:
Reactants were added into 50 ml RBF and rfx for 4 hours in oil bath T 90-100 C, in the beginning gently swirling the flask so in 15 minutes all
hydroxylamine hydrochloride dissolved and bubbles of boiling disappeared (no solids present, methanol boils silently without bubbling because of its
low b.p.).
After 2 hours there was again a solid present in the reaction (this is now the product) and the boiling bubbles observed again.
After 4 hours of reflux the reaction was cooled down to room temperature and then to +4 C (outside, winter temperature) for few hours.
Solid product sucked on cold sintered glass outside (+4 C winter temperature).
Washed 2 times with 10 ml cold water, the product a little sticky on sinter.
Washed 2 times with 5 ml of 50% methanol : water, the product nice crystalline, the stickiness lost on the first washing already.
Air dried for 1 day, during 1st half a day the weight loss was 0,04 g, during second half a day the weight stable.
Yield 0,59 g 24,5% (2,45 mmol, Molar mass 240,26 g/mol)
Original method suggests 3 hours reflux but in my experiment it seemed that at the end of 3rd hour more and more of the product was crystallized in
the flask so I decided to prolong the reflux by an extra hour from 3 to 4 hours totally. Maybe it should be investigated whether it was only my false
feeling or whether it is worth to do it to improve yield. My goal was just to prepare a little of this Niclon indicator and create a color complex
with Ni2+
I do not know whether stirring or boiling chips are necessary, I did only little of sporadic manual swirling at the beginning and did not use boiling
chips (in the middle of reaction methanol boiled smoothly and at the end there was again solid product present which facilitated boiling).
No escaping HCl observed.
Top of condenser was loosely closed with a stopper to prevent entering unnecessary air and thus moisture and allowing back reaction hydrolysis
(original method does not do it, but by my opinion the less water then less back hydrolysis of the oxime to ketone in HCl environment - I already
hydrolysed oxime to carvone in even less acidic environment in quite diluted oxalic acid in water).
Another method with much more complicated derivate used 3 fold excess of hydroxylamine hydrochloride with anhydrous K2CO3 and 80 hours reflux (1 mmol
substrate + 6 mmol hydroxylamine hydrochloride + 3 mmol K2CO3) - see the attached file IJCB 38B(10) 1208-1210.pdf
Unlike yellow reactant benzil the product diphenylglyoxime is white. Maybe the methanolic mother liquor could be further reacted with extra
hydroxylamine and K2CO3 to produce more of the desired product?
some links:
I could buy this Niclon indicator for 10 EUR per 10 g but then there is less fun, isn't it?
https://aukro.cz/indikator-niklon-p-a-diphenylglyoxime-c14h1...
 
https://sci-hub.ee/10.1139/v75-043
copy/pasted useful information:
A 0.02% alcohol or acetone solution of
a-benzildioxime which has been made somewhat amoniacal
will immediately produce a copious intensely red precipitate
of C2 H22N4O4Ni with a solution containing
0.002 mg. nickel in 5 cc. (~ part in 2,000,000). Ten
cc. of an ammoniacal solution containing 1 part per
1,000,000 of nickel and 100 times that amount of
cobalt gives a distinct and immediate test for nickel.
Iron, silver, magnesium, chromium, and manganese do not
interfere with the qualitative determination. Large
amounts of nitrates seriously affect the determination
and must be removed by evaporating with sulfuric acid
before adding the reagent. In the quantitative determinations
amounts containing less than 0.025 g. nickel
should be used. A slight excess of a warm solution of
the reagent in alcohol, to which ammonia has been
added, is added with stirring to the ammoniacal nickel
solution and warmed for a few moments on the water
bath.
Attachment: IJCB 38B(10) 1208-1210.pdf (597kB)
This file has been downloaded 290 times
Attachment: v75-043.pdf (177kB)
This file has been downloaded 283 times
now photos:
reactants, note the pale yellow color of benzil
 
reactants in the flask and putting the flask into oil bath
 
methanol boiled with bubbles only at the beginning (first 15 minutes) until hydroxylamine hydrochloride dissolved completely and also for the last 2
hours when the product crystallized out
 
after 4 hours the reflux was done, white crystals of product visible, on cooling down more crystals formed especially on the surface which then sank
to the bottom

after cooling down the product was sucked on sinter, washed with water and 1:1 methanol:water
     
air dried product, note the white color unlike pale yellow reactant

complex with Ni2+ very very very diluted, the indicator was dissolved in warm methanol, in the attached file v75-043.pdf they used dimethylsulfoxide
as a solvent
 
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
IR looks pretty okayish in OPs post, just very weak signal.
https://sdbs.db.aist.go.jp/sdbs/cgi-bin/IMG.cgi?imgdir=ir&am...
|
|
|
|









