| Pages:
1
2 |
novicechemstudent
Harmless

Posts: 1
Registered: 2-4-2016
Member Is Offline
Mood: No Mood
|
|
Drying Acetic Acid?
Can Acetic Acid be dried by fractional distillation? Will Acetic Acid forms an azeotrope with water?
If not, Can I dry acetic acid by first adding sodium carbonate to convert all acetic acid to sodium acetate, then distill the water off , then add
concentrated sulphuric acid back to sodium acetate to convert back to acetic acid then distill the dry/glacial acetic acid by distillation?
|
|
|
Sulaiman
International Hazard
Posts: 3721
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
as there are current acetic anhydride discussions you should read those first,
especially with 'student' as part of your username 
|
|
|
semesa
Harmless
Posts: 21
Registered: 13-10-2015
Member Is Offline
Mood: No Mood
|
|
It doesn't form an azeotrope but separation by fractional distillation is very impractical(impossible for all intents and purposes in amateur lab.)
Threads without a reference/s should be posted in beginners forum as per rules.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Quote: Originally posted by Sulaiman  | as there are current acetic anhydride discussions you should read those first,
especially with 'student' as part of your username |
Fate tempted, and as happened to me with this question some time ago, instant Karma.
glacial acetic acid and acetic anhydride are two distinctly separate compounds.
Edit:
Glacial acetic acid from vinegar is a project i have on the go right now, in order to make acetic anhydride.
React your vinegar (preferrably clear) with sodium bicarbonate.
Boil down substantially and collect some crystals.
Recrystallise just for the fun of it.
Dry the sodium acetate in an oven at 150C for an hour or two.
Crush up the pretty dry sodium acetate and distill with conc sulphuric acid.
What 'comes over' is glacial acetic acid (a.k.a. GAA), or as near as damnit.
Personally i want more GAA to make zinc acetate which hopefully will decompose to acetic anhydride when distilled under vacuum.
At this moment it's just a pile of dilute sodium acetate in a massive pan on top of a barbecue ... just 10L of water to boil off 
Then again, worth it for over a kilo of sodium acetate
[Edited on 2-4-2016 by aga]
|
|
|
Homelabchemist
Harmless
Posts: 4
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
first fractionally distilling and then drying over magnesiumsulfate.
That is what i would do.
|
|
|
Sulaiman
International Hazard
Posts: 3721
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
I thought
anhydrous + H2O = glacial
and much of the other info. in the anhydride threads is relevant.
BUT you are correct in assuming a level of fuzzy thinking on my part, sorry for that.
since I have the anhydride I've not tried to dry dilute to glacial
I do not want to buy any mode anhydride
so I too need to dehydrate some acetic acid to glacial soon
learning .... slowly
I'll be quiet now
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
I once thought that glacial acetic (having no water) was acetic anhydride.
Makes logical sense, as far as the words go, which is why i once said exactly the same thing as you did.
Someone here on SM corrected me, which is the only reason i now know different, as you now know different.
Do not be quiet : say things, and flaws such as this are discovered and corrected, perhaps flaws on Others' rather than your own knowledge - you never
can tell.
Nothing Ventured, Nothing Gained ...
Outcome = knowledge++ either way.
|
|
|
teodor
National Hazard
Posts: 922
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
I'm doing experiments with drying acetic acid just now. I found a useful article:
https://www.researchgate.net/publication/287217410_Investiga... There are 3 basic methods studied there with a lot of references.
Now I try to use a Widmer column to get an idea of how a usual fractionation goes. There is some interesting observation of condensation properties
depending on a water percent. Even a small percentage of water increases the surface tension of condensate, so the presence of water is clearly
visible because in this case, the condensate forms many drops on a glass surface. And water-free acid doesn't.
I think this graph is useful:
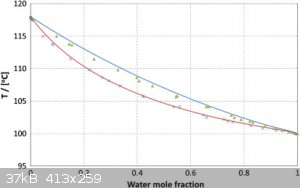
[Edited on 24-11-2021 by teodor]
Update: the reference to the article was fixed.
[Edited on 24-11-2021 by teodor]
So, with a Widmer column (250mm overall with 160mm the spiral part), I've got 2/3 of the volume distilled 102-117C and 1/3 117-118C. Let's take a
precision here as +-1C (the thermometer I use was not calibrated in the range 100C+ yet)
[Edited on 24-11-2021 by teodor]
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi teodor, when you achieve quite concentrated acetic acid with only few % of water then the convenient method to further concentrate it is by
freezing/crystallization where crystals are enriched in acid and the liquor enriched in water. I tried that with 2 bottles of 20 years old commercial
98% acetic acid, I let them in my fridge at 4 C, then just decanted the liquid phase and then melted the crystals back into liquid glacial acetic
acid.
Determining melting / freezing point is quite sensitive test to tell you how much concentrated is the acid when it is close to 100%, there are tables
available for concentration of the acid and its freezing point.
Anyway the column distillation is very good skill in chemistry. With the column did you also use a head to adjust the reflux ratio (how much drops to
take and how much to return back into the column) ?
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I once ruined a reaction because I thought what I had was absolute GAA and I was impatient and didn't let the whole bottle thaw. I used the first bit
off melt, which wasn't GAA. The second try did work.
[Edited on 24-11-2021 by Tsjerk]
|
|
|
teodor
National Hazard
Posts: 922
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
| Quote: Originally posted by Fery |
Anyway the column distillation is very good skill in chemistry. With the column did you also use a head to adjust the reflux ratio (how much drops to
take and how much to return back into the column) ? |
Hi Fery. Exactly, I am doing this for getting new good skills.
I tried to find the special head which allows adjusting the reflux ratio but those I saw cost something like 500-700 EUR. I think it is possible to
find just a cold finger and use it with a normal claisen adapter, but I didn't find the matched parts yet. Probably I can try to use also a condenser
instead of a cold finger. But I'd like to read about other people working experience with such adapters before trying it.
I had some issues with precise temperature measurement but now I bought several mercury thermometers and the mercury thermometers are fast enough to
catch exactly the fraction I need. Also, they show when the column is not working properly (the column of mercury is jumping in this case). With
proper heat adjustment based on this effect, I now can get a quite good reflux ratio by using a 2-neck claisen adapter like on the foto. Because of
the second neck, it returns more to the column than allow to pass through - the effect I use.

|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi teodor, which is the size of your ground glass joints on the column you used?
I have 2 types of variable reflux ratio distillation heads:
[0]
https://www.sciencemadness.org/whisper/files.php?pid=658651&...
https://www.sciencemadness.org/whisper/files.php?pid=658651&...
[1]
https://www.sciencemadness.org/whisper/files.php?pid=662094&...
https://www.sciencemadness.org/whisper/files.php?pid=662094&...
I used the type [0] with 1 m long 30 mm inner diameter Hempel column packed with Raschig rings (circa 7 x 7 mm) to distill ethanol from 80% to
azeotrope, it was very powerful, the methanol content below 0,1% and also all other contaminants together below 0,1%. My friend Bedlasky performed a
gas chromagraphy in his job. He told me that they have never had such pure ethanol in his job for syntheses (they produce diethyl oxalate among a lot
of other products). I did it by fermenting sugarbeet sugar from supermarket + water + crushed apples (abandoned apples orchard), then 2 simple
distillations (from 10% to 40%, then from 40% to 80%) and finally the column distillation from 80% to azeotrope. This head was powerful enough to cool
a vapor from 4 L flask in 750 W heating mantle.
The type [1] is designed especially for vacuum distillations (but could be also used for atmospheric pressure distillations) and I used it for
separation of orto nitrotoluene, this was not too much successful, my product still contained something like 10-15% of para isomer, Bedlasky did a gas
chromatography with it too (they produce nitrobenzene in his job too so he had also samples of analytical grade nitrotoluenes to calibrate GC). I
think I used too wide column (30 mm) and using narrower could make separation more efficient (the back flow should be slightly below column flooding
then the separation is the best which I did not achieve in my case as the back flow was only 2-3 drops per 1 second in comparison with ethanol where
so much drops that I was unable to count them so maybe 20 drops per second, in both cases I used 30 mm inner diameter columns so in the second case I
should use 10 times smaller area of the inner diameter so something like 10 mm instead of 30 mm). The distillation flask with [1] had IIRC only 0,5 L
(so 8 times less than the ethanol distillation 4 L flask) and magnetic stirrer with heating I do not remember exactly maybe 250-350 W but extra loses
due to using oil bath in aluminium pot.
I got some extra heads of type [1] but all of them are used (dust on outside could be cleaned easily and little of precipitated salts inside from
circulating water could be dissolved in acid or could let it be as that does not affect the functionality). If someone needs such a glass I can share
(only the type [1] which I have in abundance). They have ground glass joints 29/32 on input and 14/23 on output. I have some adapters / reductions
29/32 <-> 14/23 (in abundance too). You need a thermometer with 14/23 ground glass joint (I can't share these thermometers as I do not have
excess of them), stem length 40 mm from the top of ground glass joint to the tip of bulb of the thermometer, slightly longer stems do not mind and fit
there too. I have never send any parcel abroad yet, but perhaps DHL could operate in whole EU?
[Edited on 25-11-2021 by Fery]
|
|
|
teodor
National Hazard
Posts: 922
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
Hi Fery.
I use 14/23 and 29/32, depending on the scale (the volume of the liquid). My Widmer column has 14/23 input/output. I plan to buy a Hempel column with
29/32 output. Because I have different interests and a lot of equipment for chemistry which I plan to buy also for the experiments which I already
have planned, it is not in my nearest shop list unless you know where to buy some big column (like that one on your photos) for a good price. I use
adapters 29/32 <-> 14/23, but I think I don't have them in abundance
As for thermometers, I use 3 types:
1. Those which fit in 14/23 adapters
2. Those which don't fit in 14/23 adapter but I have 29/32 thermometer & claisen adapter for them (they are high precision mercury thermometers
with 0.1C and 0.01C scale)
3. Thermometers with 14/23 ground glass joint. I have the source of them so I can get them in abundance But I prefer to use adapters for cases that don't require a vacuum because I can better adjust the position of the
bulb.
0.1% of methanol in the distillate is really cool. And a lot of chemicals I buy really require purification (and particularly GAA is very hygroscopic
and probably requires dehydration in many cases before usage).
Thank you for the proposition. Unless I will have a big Hempel column, [1] looks a bit bulky for my current setup. But I will think about it.
As for practical application regarding GAA drying, I think the method of water azeotrope with acetate ester (e.g., ethyl acetate) is a good one that
could be very effective even with a small column.
It is good to hear that you and Bedlasky are friends.
And yes, DHL can operate in EU. And DPD also. [1] probably is not easy to pack in a box. But there is another possible option - I am travelling a lot
by car 
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
OK then I try to send you the distillation heads plus reduction adapter. The reduction adapter is this type:
https://www.verkon.cz/vlozka-prechodova-redukce-obracena-1/
You'll have to put the 14/23 of the reduction adapter into your 14/23 column and the wider part of the adapter 29/32 is where to insert the 29/32 male
joint of the distillation head.
I received a lot of parcels including on wooden pallets but I have never send any parcel abroad. Maybe I should learn new skills. If you are with your
car in CZ I will give you that heads personally. I have also a lot of reagents including glacial acetic acid. I converted whole house into lab, there
is only 1 room without any equipment/glass/reagent and that is sleeping room, everything else is packed with chemistry.
From my experience 1 cm inner diameter column should be suitable for distillation flask like 500 ml.
30 mm inner diameter column for 4 L distillation flask, but that also depends on the power of your heating mantle.
The reflux ratio on distillation head should be adjusted to something like 10 drops back into the column and 1 drop into receiving flask. The back
flow should be slightly below flooding the column, this is adjusted by first flooding the column and then slightly reducing the power of heating
mantle so the flooding disappears. The bigger flow the better separation (but not flooding, flooding is bad situation which destabilizes column
equilibrium).
Yes less than 0,1% of methanol from apple cider is quite good as apples are high on pectins which are source of methanol.
|
|
|
Mixe
Harmless
Posts: 41
Registered: 2-6-2017
Member Is Offline
Mood: No Mood
|
|
But ... is distillation really necessary? Can't you just freeze it dry?
|
|
|
SWIM
National Hazard
Posts: 970
Registered: 3-9-2017
Member Is Offline
|
|
Freezing would be easier.
Sulfuric acid plus a dry acetate easier still.
I think teodore is doing this partly for the challenge and partly to gain experience with difficult distillations.
Acetic acid is good for practice since it is easy to assay the results and it's cheap to buy.
Been thinking of trying this myself just to see how hard and time consuming it is.
Nice Widmer column, BTW. Is that like 400 mm?
I am very curious as to how well those Widmers work as I have a couple but they're missing their central spirals and I'm not sure if it's worth
getting new glass spirals made.
For those unfamiliar with Widmer columns, the vapors travel up to the top of the column, back down to the bottom through an inner concentric column,
and then back up to the top through an even smaller concentric tube which contains a glass spiral like a Dufton column.
His also appears to have an outer insulating jacket.
[Edited on 6-12-2021 by SWIM]
|
|
|
teodor
National Hazard
Posts: 922
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
Hi SWIM.
Yes, I am doing it mainly to understand fraction distillation. I think it is a very important technique in organic chemistry. Actually, I was in a
process of making ethyl acetate by Fischer esterification. And then I started to separate different azeotropes formed to check the amount of water
that was generated. I used absolute ethanol so I was surprised why I got so much water. And then I discovered that most came from acetic acid which
was "glacial" but not perfectly dry. So, actually, there was no point to use absolute ethanol with the wet acid ...
Also, freezing acid and getting the pure product is a longer process. And with distillation when you get the sharp boiling point of the pure acid you
can just replace a receiving flask.
About the column: 250mm overall with 160mm the spiral part. It has an outstanding jacket. I had it without a spiral also and bought the spiral as a
separate part from the same supplier.
The performance is not so great but one day probably I will be able to compare it with Hempel and then I probably I will be able to give some numbers.
Now I try it in different modes with different vapor speeds, returning something back, etc. In the course of it, I did some interesting observations
but I plan to write something about it later.
I use it to purify different alcohols and different solvents and it is really helpful - sometimes I see the sharp difference between fractions (from
chemical pure sec-butanol I've got 3 fractions with different smells). And it is really fascinating to observe how different layers of the Widmer
column work and how the liquid goes down through the central part in a spiral movement.
|
|
|
theAngryLittleBunny
Hazard to Others
Posts: 130
Registered: 7-3-2017
Location: Austria
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by novicechemstudent | Can Acetic Acid be dried by fractional distillation? Will Acetic Acid forms an azeotrope with water?
If not, Can I dry acetic acid by first adding sodium carbonate to convert all acetic acid to sodium acetate, then distill the water off , then add
concentrated sulphuric acid back to sodium acetate to convert back to acetic acid then distill the dry/glacial acetic acid by distillation?
|
That way you'll get about 95% acetic acid since you're using only 95% sulfuric acid, you could get 99% by using an excess of sulfuric acid to absorb
any water though.
I distilled propionic acid from a mix of sodium propionate and concentrated sulfuric acid, I probably used maybe 5% excess of the latter. Even after
drying the propionic acid with Na2SO4 after distillation it still seemed to contain a lot of water. I tried to make propionyl bromide by adding PBr3
and the yields often were awful, like 30% and it fumed from all the HBr that was produced. But I think something like 20% excess H2SO4 would keep any
water from distilling over. To be clear, I used 1 mole of H2SO4 per mole of sodium propionate, using 0.5 moles is theoretically possible but usually
gives poorer results and makes the distillation a pain.
|
|
|
teodor
National Hazard
Posts: 922
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
Sodium acetate is a trihydrate. I think that drying sodium acetate and sulfuric acid is not so trivial.
If you can get the anhydrous sodium acetate the best way to go is probably to make acetic anhydride which can be used for drying acetic as well as
sulfuric acid. At least in some parts of the world (EU) acetyl chloride is more readily available than 99% sulfuric acid.
Also 99% sulfuric acid and perfectly dry sodium acetate (which is a quite temporary thing) still contain 1% of water which is already comparable to
our typical glacial acetic acid which we try to dry. And I am not sure that sulfuric acid has a bigger affinity to water than acetic acid - in any
case, this is hard to check - the mixture of both is carbonized on heating.
And about getting perfectly dry acetic acid, from "Comprehensive inorganic chemistry" (Sneed/Brasted/Pray):
".. the liquid is not easy to free of water; simple distillation or fusion is ineffective, allowing up to a half a per cent of water to remain. ... It
is necessary to distill the liquid under reduced pressure in the presence of P2O5, and then (!) distill and freeze alternately, in order to obtain a
product of the highest purity" (the reference given is J. Timmermans, "Physico-chemical Constants of Pure Organic Compounds, Elsevier 1950).
[Edited on 7-12-2021 by teodor]
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
theAngryLittleBunny wasn't your Na propionate some form of hydrate instead of anhydrous?
teodor - of course increase your skills in column distillation, but I will give you also 1x bottle of commercial concentrated acetic acid :-)
in home lab the most trivial way do determine concentration of glacial acetic acid is to let it to crystallize in fridge at +4C and then determine its
melting point in the bottle and lookup into tables... but all bottles with commercial acetic acid I tried always become supersaturated so it was
necessary to let them in the fridge or cold room for a long time (I just want to say that if no crystals after 1 day it still could be very
concentrated acetic acid, you must patiently wait, if you want to produce larger crystals then let the bottle with crystals to almost melt at room
temperature and when only little of crystals in the bottle then put it into fridge again or cold room if you think something like +10 C would be OK)
|
|
|
teodor
National Hazard
Posts: 922
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
| Quote: Originally posted by Fery | | but all bottles with commercial acetic acid I tried always become supersaturated so it was necessary to let them in the fridge or cold room for a long
time |
Good point, Fery. I also had such a bottle with the label saying "for analysis, 99.5%". Never saw any crystals in a cold room. Also, there are some
concentrations that are still a liquid at -18C after a day. But then I redistilled all those scum through a column and now it freezes in less than an
hour (I believe after several minutes) at -18C. But it thaw very slow starting from outside, so I have no idea at which moment I should measure the
temperature.
[Edited on 7-12-2021 by teodor]
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Teodor, you can measure by inserting thermometer into the liquid portion of the acid when there are only few crystals (something like 90% already
liquid and 10% crystals). If there is only little of liquid and almost everything is crystalline at that moment the crystals may be almost 100% acid
and the liquid part could be acetic acid with most of water present in the bottle (liquid portion much more diluted than the almost pure crystals).
Acetic acid is very cheap, 5 L canister 99,5% p.a. = G.R. = for analysis for 16 eur
https://sklep-chemland.pl/en/odczynniki-chemiczne/cz-d-a/kwa...
I usually do not distill commercial reagents / solvents as that is only waste of my time but it's good to have this skill in column distillation. I
distill only things which I produce. E.g. I produced ethanol by myself as I was angry because of methyl ethyl ketone contamination so I fermented
sugar and then redistilled twice simple distill to 80% concentration and for the 3rd time column distill to azeotrope. For some reactions I need
ketone free ethanol, and yes I know about NaOH aldol condensation of MEK to remove it, but whenever possible I prefer methanol or isopropanol. I hate
this shitty MEK added into commercial ethanol. Sorry for out topic.
When I was small boy I of course tried to distill diluted acetic acid to concentrate it. I made wine by fermentation (circa 10% ethanol
concentration), then opened the bottle and let acetic acid bacteria to perform fermentation and then distilled. But that old time I did not have
equipment to determine the final concentration of the acid obtained. When I was student I worked during summer holiday in a factory producing vinegar,
they produced also alcoholic beverages and part of the distilled ethanol from fermentation of molasses was transported through tubes into building
where it was diluted and fermented into acetic acid, the fermentation tanks were huge and full of wooden chippings, liquids flowing down from top to
bottom and air from bottom to up, the vinegar was finally diluted to 8% and bottled, but that's another story.
|
|
|
teodor
National Hazard
Posts: 922
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
Fery, I tested the acid I've purified by distillation through my column and it has a temperature of the liquid 16.2C and the temperature inside ice
15.7C. I waited almost the full day but it only thaws a bit, so most of it is still one piece of ice. Which table I can use to check the
concentration?
The rest which was distilled as a water-acid mixture was half-frozen after 1.5 hr and had a temperature of the liquid 9.2C. I decanted the liquid.
My column gave me nearly 50/50 of the water acid (m.p 9.2C +) and the acid with m.p. 16.2C, but I don't know the starting concentration, it was mostly
a residue of esterification.
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi teodor, this is almost pure acetic acid (m.p. 16,6 C).
I found some info here, see the last table at the bottom of the page:
http://www.ddbst.com/en/EED/SLE/SLE%20Acetic%20acid%3BWater....
T 285.11 K mole fraction of acetic acid=0.90600
T 289.83 K mole fraction of acetic acid=1.00000
also the last picture on the page
http://www.ddbst.com/en/EED/SLE/Images/SLE%20Acetic%20acid;W...
90,6% (molar mass) acetic acid has its solid-liquid equilibrium point 4,72 C lower than 100% acid. This part of the curve is almost linear, for our
home chemistry purposes you can use linear approximation in this part.
|
|
|
garphield
Hazard to Self
Posts: 58
Registered: 9-12-2019
Member Is Offline
|
|
Could you just add some sulfuric acid to the wet acetic acid before distilling to retain the water? A 80% solution of sulfuric acid has a boiling
point of approximately 200 C, which is much higher than the boiling point of pure acetic acid and should therefore be fairly easy to retain in the
boiling flask. Not useful for concentrating dilute acetic acid, but if you wanted to remove the last bit of water from glacial acetic acid it should
work, unless there's some acetic acid-sulfuric acid azeotrope or something I don't know about.
|
|
|
| Pages:
1
2 |










{kind=link}