| Pages:
1
2 |
Assured Fish
Hazard to Others

Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
| Quote: |
Tosylation of the alcohol followed by treatment with LDA to give the target compound via an intramolecular alkylation.
|
I knew it!
Im not crazy, there is a way to do that. Mwahahahahaha
|
|
|
j_sum1
Administrator
Posts: 6333
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
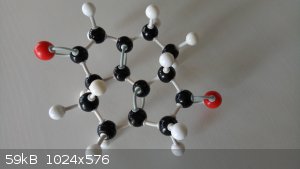
I made it! I made it!!
Yield is a bit bleak though. I only got one molecule.
|
|
|
j_sum1
Administrator
Posts: 6333
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
On a more serious note...
Some things that may or may not have been noticed by those working on this problem.
1. There are two chiral centres and so two diastereomers. (Labelled in the following photos by the purple bonds.)
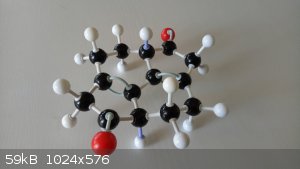 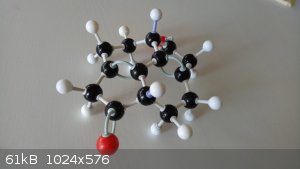
2. There seems to be a lot of bond strain. At least if the model kit is anything of an indicator. The structure really wants to fly apart. The
trans isomer in particular. I am not predicting that the synthesis will be easy at all.
[Edited on 29-9-2018 by j_sum1]
|
|
|
j_sum1
Administrator
Posts: 6333
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
Throwing it into my phone app...
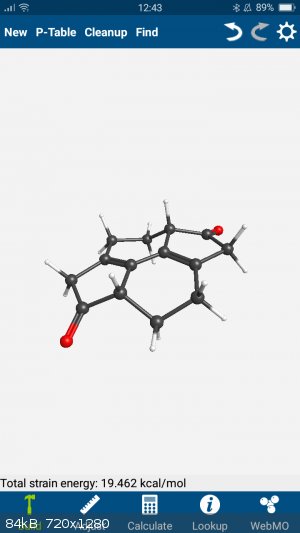 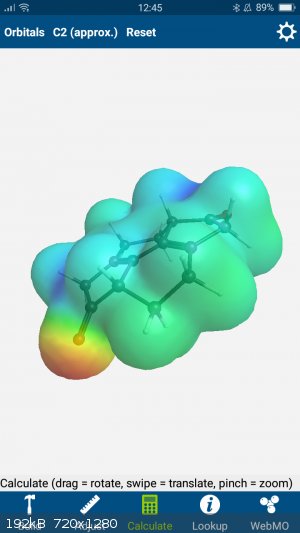
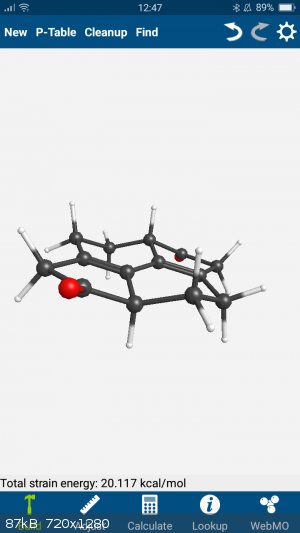 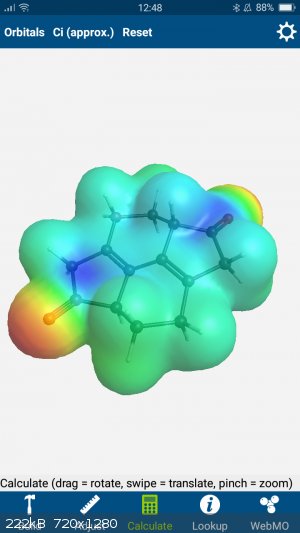
(Webmo if anyone is interested.)
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Assured Fish  | | is it possible for an alcohol to react in an aldol condensation with an enol instead of classically reacting an aldehyde with an enol?
|
I think this was the reaction you were hoping to find and are so ecstatic about now.But this is merely the
alkylation of enolates using an activated alcohol(tosylate in this case) as the electrophile.You can't call this an aldol condensation. actually,there are 5 chiral centres.But since
jackson wants to use this molecule as a base for bucky balls,I don't think stereochemistry would matter much.As long as it doesn't have to fit in a
particular receptor(like drugs),it should be fine.I may be wrong though 
Also,the synthesis is already very hard as it is.Trying to make it asymmetric would be next to impossible.
I suggest we try getting a product first as proof of concept.This could be done by selecting the 2 best routes posted,correcting their flaws and
optimising them( like figuring out how many steps could be run in one pot).Then jackson could do a trial run and report back here.
[Edited on 29-9-2018 by CuReUS]
|
|
|
j_sum1
Administrator
Posts: 6333
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
| Quote: | | actually,there are 5 chiral centres. |
Then either I can't count or I have some confusion over what a chiral centre is. I am happy to be educated.
My point is that playing with the model kit I get two isomers: one concave and the other somewhat twisted. Both structures are quite strained to the
point that the kit pops open. (I know this is no substitute for good theory and supporting calculations. But modelling does help with getting a feel
for things.)
[Edited on 29-9-2018 by j_sum1]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
While trying to come up with the route I suggested in the previous page,I faced a dilemma about which protecting group to use.I was torn between
converting the primary alcohol and enol to an ester(benzoate) or ether(methyl ether).In the end,I chose benzoate since it was less toxic and the
deprotection was a breeze.
But that time I had a nagging doubt - would the Mg used for the pinacol coupling react with the ester ?
I went through greene's book on protective groups,and I am sorry to say, my fellow chemists,but my doubts have been confirmed  - https://www.sciencedirect.com/science/article/pii/S004040390... - https://www.sciencedirect.com/science/article/pii/S004040390...
https://www.sciencedirect.com/science/article/pii/0040403995... (don't know why they published the same thing again after 2 years using different
authors)
Although the deprotection condition is Mg/MeOH,rt for 13 h,I don't want to take any chances
So I suggest we proceed via the ether route.The reaction stays the same,the only difference is that the starting material is different ( CAS no:
4683-50-5) and we do a methylation step instead of benzoylation.
the methylation could be done with dimethyl sulphate/Barium hydroxide-https://pubs.acs.org/doi/10.1021/jo00374a020
or methyl iodide/silver oxide-https://pubs.acs.org/doi/10.1021/ja00545a041
deprotection can be done by trimethyl silyl iodide -https://pubs.acs.org/doi/10.1021/jo00443a033
or BBr3-https://www.jstage.jst.go.jp/article/bbb1961/42/1/42_1_131/_...
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Ive been researching how one could go about preparing the starting material 3-hydroxy-2-cyclopentenone or an ether thereof.
I found the following paper, which looks promising although entails 3 steps.
https://cdn-pubs.acs.org/doi/10.1021/jo010593b
Sadly ive yet to get access to see what its limitations are or the exact conditions used but it would mean starting from commercially available
acrylic acid acid or an ester of acrylic acid, acetylene, HBr and a ruthenium catalyst of some kind, i suspect the solvent would be THF.
Not sure which is looking more promising at the moment, the naphthalene route or the cyclopentenone route but the later is certainly more developed.
Although some of the reagents are a tad exotic.
[Edited on 1-10-2018 by Assured Fish]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
There is no need to make it,since jackson can directly buy it. 
| Quote: | | Although some of the reagents are a tad exotic. |
They might be "exotic" from a home chemist's point of
view,but they are quite commom in an university setting,where jackson is working. 
[Edited on 1-10-2018 by CuReUS]
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
Just to clear things up im not working at a univeristy currently so these reageants are going to be difficult to get access to. I know people that
work at a university that can help me get access to analytical equipment. I do know someone that could potentially help me get access to chemical
suppliers .
|
|
|
TGSpecialist1
Hazard to Self
Posts: 53
Registered: 24-12-2017
Member Is Offline
Mood: always tired
|
|
You will probably enjoy this paper:
https://sci-hub.tw/10.1002/anie.200705775
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
the reason I said "working" was because I thought you
are a student who has access to an university lab and reagents since you said you wanted to make bucky balls,and another member had also worked with
them when he was in school.(zts16 I think) | Quote: | | I do know someone that could potentially help me get access to chemical suppliers . |
You could also buy them
online cheaply,from chinese suppliers.See the "Reagents and apparatus acquisition" sub forum for more info. what's your point ?
strained compounds exist.That doesn't mean they can't be synthesised -https://en.wikipedia.org/wiki/Bicyclobutane
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
Im an Idiot.
I forgot to ask a very important question.
This molecule is a segment of a larger molecule. I forgot to ask if the larger molecule could even be made from this molecule.
I will attach a photo of a model of the full molecule.
P.S. the red bonds are single bonds, white bonds are double bonds. The open double bonds on the end are supposed to be where the oxygen it bonded.

|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
The precursor molecule could be made by the gringard reaction of
4-amino-2-bromomagnesium-but-1-ene and 3-amino-acrolein. Then this could be cyclized by a lewis acid.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Holly jebus.
I dont have the time to answer your question in full but primary and secondary amines are incompatible with grignard reagents as the organo magnesium
halide carbon will just deprotonate the amine.
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
Could the amines be converted into a different group?
If so, would something like a nitrate group be fine?
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
| Quote: |
Could the amines be converted into a different group? If so, would something like a nitrate group be fine?
|
I think you mean a nitrite group, organo nitrates are very unstable and dangerous to prepare e.g. nitroglycerin.
The answer is no, you need to do some research on the exact mechanism of a grignard reaction so you can understand its limitations.
Another group is possible yes but your route is flawed i have a better one but will need to do some work on it which will take a few days (im very
busy at the moment).
Now, as for your fullerene material, I need to crush your expectations.
You cannot make that compound starting from the pyracenedione species we have been working on in this thread.
A method for making such a compound is probably well beyond what the members on this site are capable of, even if we were to come up with a
theoretical route, the route would likely involve so many steps that the sheer difficulty in preparing the compound would make it synthetically
useless.
It certainly looks like it might have some uses to the species but these kind of projects are the kinda things large research labs spend tens of
millions of dollars on and several decades to develop, usually in the hopes of earning some Nobel prize or something.
The major issue with preparing it is trying to prepare a large 16 carbon substituted cyclocarbon species, which would likely be the backbone for the
material.
Im sure i could find some approach to preparing such a molecule (with many flaws) but you will likely never be able to make it.
Im sceptical of you being able to prepare the hexahydo pyracenedione molecule as is.
Buckyballs or buckminsterfullerene is not prepared using methods commonly used in organic chemistry (that would be insane), it is by some miracle
prepared by simply passing an arc between two graphite electrodes at a very high voltage and then isolated using an organic solvent as the fullerene
is soluble whereas carbon is not.
This kinda information is available from the video periodic videos did on the substance.
https://www.youtube.com/watch?v=ljF5QhD5hnI
If you want to prepare it then by all means, it would be an interesting project and this paper may be of some use to you.
Beyond that though, i can do some research about preparing that thing of yours, i will make no promises, i will almost certainly fail and if i come up
with something it will be a mess.
Attachment: zhu1993.pdf (317kB)
This file has been downloaded 377 times
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
I actualy found a paper that created C60 using a chemical process.
I will send a link to this thread if I can find it.
My analogy to a buckball for my final molecule was not a very good one as the final molecule I wanted to make is not a fullerene. This was more of a
theoretical idea than something I was going to practically pursue. I will probably attempt synthesis of the pyracenedione once I get some more
materials and equipment needed for it.
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
http://science.sciencemag.org/content/295/5559/1500.full
Here is the link to the article.
|
|
|
SWIM
National Hazard
Posts: 970
Registered: 3-9-2017
Member Is Offline
|
|
I sure can't see 5 chiral centers in that molecule either.
Is there some stuff about heterocyclic structures or something I'm forgetting here?
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
I also do not see five chiral centres, havent a clue why Atara stated that, perhaps an oversight on his part.
Anyway, this is route for the preparation of the cyclopentenone species.
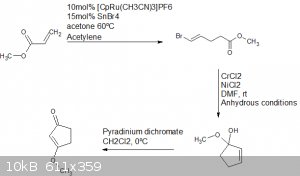
Based on the research done in the paper trost2001.
for each of the 3 steps they worked up by way of ethyl ether/petroleum ether over silica gel by way of chromatography.
The last step needs to be done quickly as the geminal diol ether compound is likely unstable, it is also moisture sensitive and chromium 2 chloride is
hydroscopic.
This is the ruthenium catalyst used in the first step.
https://www.sigmaaldrich.com/catalog/product/aldrich/667412?...
Yields would likely vary too much to be of much importance but for each step they reported above 60% and the 3rd above 80%.
It would be better if you read the paper yourself.
That first step is somewhat problematic as acetylene is probably not highly soluble in acetone at the reflux temp required for the reaction, a
continuous flow of acetylene would likely be required, which would sap energy away from the reaction so a strong heat source would be a necessity.
Thankfully an excess of acetylene would not hinder the reaction or take place in side reactions.
Unfortunately there is nothing easy about this kinda synthesis, non of the materials with exception of the solvents are easily accessible.
Honestly the more i read into it the more i start to learn towards Atara's route starting from naphthalene.
All of this stuff is just madness, well beyond what i would be willing to do, especially with no obvious use for the end product.
A hell of a lot of money would be required to buy reagents, or time required to prepare the more easily prepped ones like PDC and nickel dichloride.
Stannic bromide would be horribly difficult to prepare, buying it would be necessary which would also be challenging.
Attachment: trost2001.pdf (143kB)
This file has been downloaded 408 times
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
atara didn't state that,I did 
And I was wrong,its 2 only
| Quote: | | Unfortunately there is nothing easy about this kinda synthesis, non of the materials with exception of the solvents are easily accessible.
|
that's because you have yourself complicated it.
I said so many times that the starting compound is directly available commercially.
Still, you insist on synthesising it,that too via a ridiculously convoluted route,when it can be made in 1 step from 1,3-cyclopentanedione(another
commercially available product) -https://pubs.acs.org/doi/10.1021/jo801911s(pg 9 of pdf,compound 74) , which in turn can be made from methyl/ethyl levulinate -https://patents.google.com/patent/US7897802B2/en
ethyl levulinate is available as biofuel - https://en.wikipedia.org/wiki/Ethyl_levulinate, or if you truly wanted to make it from scratch,you could make it from sugar -https://en.wikipedia.org/wiki/Levulinic_acid#Synthesis
| Quote: | | Honestly the more i read into it the more i start to learn towards Atara's route starting from naphthalene. |
But the birch reduction step is doubtful,and there is no ref to back it up.
the only reason I suggested the cyclopentanone route was because I couldn't find a ref for the reduction of PCD,which I still think is the easiest way
to make this molecule.
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
Another idea for a route could be:
Synthesis of 1,5 dimethyl napthalene as shown here: https://wikivisually.com/wiki/2%2C6-Dimethylnaphthalene (without the final step)
Then bromination of both of the methyl groups to form 1,5 dibromomethyl napthalene
Then reacting the 1,5 dibromomethyl napthalene with sodium cyanide to form 1,5 diacetonitrile napthalene
Then hydrolysis of 1,5 diacetonitrile napthalene to form 1,5 napthalenediacetic acid
Then conversion of the OH groups in the acetic acid to H by chlorinating or brominating them and then replace the Chlorine/Bromine with Hydrogen using
H2/Pd in the presence of BaSO4.
Then it could be cyclized with H2SO4 to form the molecule
|
|
|
clearly_not_atara
International Hazard
Posts: 2799
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
| Quote: | | I also do not see five chiral centres, havent a clue why Atara stated that, perhaps an oversight on his part. |
I didn't say anything about five chiral centers or for that matter anything about chirality at all.
I'm not sure about the Birch reduction of 1,5-naphthalenediols. I can't find anything like it in the literature. Most general references tell me that
usually only one ring of naphthalene is reduced, though. That alone suggests that the method needs to be revised a little. Or maybe it's just a
stoichiometry thing.
But the perhydrogenation of that naphthalene ought to be achievable somehow. What's less clear is what we end up with afterwards, and how to turn that
into what we want.
|
|
|
Outer
Harmless
Posts: 38
Registered: 24-11-2008
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Jackson | Another idea for a route could be:
Synthesis of 1,5 dimethyl napthalene as shown here: https://wikivisually.com/wiki/2%2C6-Dimethylnaphthalene (without the final step)
Then bromination of both of the methyl groups to form 1,5 dibromomethyl napthalene
Then reacting the 1,5 dibromomethyl napthalene with sodium cyanide to form 1,5 diacetonitrile napthalene
Then hydrolysis of 1,5 diacetonitrile napthalene to form 1,5 napthalenediacetic acid
Then conversion of the OH groups in the acetic acid to H by chlorinating or brominating them and then replace the Chlorine/Bromine with Hydrogen using
H2/Pd in the presence of BaSO4. |
Both products (diacid and dialdehyde) can be obtained by shorter way. 1,5-dibromomethyl napthalene can be transformed into bis-Grignard reagent
(-CH2MgBr). Its reaction with CO2 will give diacid, while reaction with HCOOEt (instead CO2) will give dialdehyde.
Cyclization of diacid (and not dialdehyde) will form diketone, but anyway it will have aromatic (naphtalene) core, while your "original" molecule has
2 double bonds in certain positions.
[Edited on 7-12-2018 by Outer]
|
|
|
| Pages:
1
2 |









