| Pages:
1
2 |
DubaiAmateurRocketry
National Hazard

Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Computational methods / calculations in Energetic materials
I recently figured out how to calculate properties of energetic materials based on several computational methods. Some of these methods are relative
of high accuracy.
Methods:
Density:
Politzer, P., Martinez, J., Murray, J. S., Concha, M. C., & Toro-Labbe, A. (2009). An electrostatic interaction correction for improved crystal
density prediction. Molecular Physics, 107(19), 2095-2101.
Heat of formation:
De Paz, J. L. G., & Ciller, J. (1993). On the Use of AMl and PM3 Methods on Energetic Compounds. Propellants, explosives, pyrotechnics, 18(1),
33-40.
Detonation performances, impact sensitivity, etc.:
Kamlet, M. J., & Jacobs, S. J. (1968). Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O
explosives. The Journal of Chemical Physics, 48(1), 23-35.
Keshavarz, M. H., Klapötke, T. M., & Sućeska, M. (2017). Energetic materials designing bench (EMDB), version 1.0. Propellants, Explosives,
Pyrotechnics, 42(8), 854-856. (This method only seems to produce semi-reliable results on detonation energy and detonation pressure)
__________________________________________________________
I have test-ran around 50 compounds already, a single computational run a Xeon quad-core computer takes about 30 minutes to 5 hours, depending on the
structure.
few month ago, DOI: 10.1002/slct.201703067
this paper published a some data on politzer's equation that predicted one CHON compound with a density of 2.10g/cm^3, and therefore, a VoD of
10,500ms, approaching the 11km/s limit.
I will perform some runs and check if its infact true, since the density calculations from politzer is usually highly accurate.
Also, if you propose any compounds that are interesting I could calculate its properties for you.
[Edited on 21-7-2018 by DubaiAmateurRocketry]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
So the first result turned in.
The claimed compound in DOI: 10.1002/slct.201703067, states that compound C1(nitro derivative) to have a density of 2.1, and a heat of formation of
1330 kJ/mol.
To reproduce the study's results, I will be copying their methods - using B3LYP at 6-31g(d,p) level, which is what they used for politzer's equation,
which is funnily, already wrong - Since politzer's paper uses B3PW91 basis set, at 6-31g(d,p).
Furthermore, using PM3, the heat of formation was calculated to be:
Gaussian 03: 622.712310698275 kJ/mol
Gaussian 09: 625.46561210138 kJ/mol
For reference, Tetrazino-tetrazine-tetraoxide or TTTO, have a heat of formation of 960 kJ/mol.
Instead of 1330 kJ/mol. This already pulls the detonation velocity from 10,500 m/s down to 9,850 m/s. While its still a very interesting molecule for
such a performance, I will complete the Politzer's density calculations soon, a computational run on this molecule may take many more hours, and I
will update this later.
The calculation still have no finished today, will update tomorrow.
[Edited on 22-7-2018 by DubaiAmateurRocketry]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
http://www.sciencemadness.org/talk/viewthread.php?tid=85962
Here I uploaded the calculated results in a new post.
|
|
|
simply RED
Hazard to Others
Posts: 209
Registered: 18-8-2005
Location: noitacoL
Member Is Offline
Mood: booM
|
|
How could you calculate density, without having an idea of the crystal structure. This is a fundamental question.
When logic and proportion have fallen sloppy dead...
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Wow, very nice! Is this based on density functional theory? Don't no much about it, though IIRC, B3LYP/B3PW91 represent a particular set of
assumptions to calculate local electron densities right? How do you know which to use for which compound? And like simply red also asks, this method
seems to use only the electron densities of the molecule itself, how does it estimate the influence of between molecule interactions? Does it also
work for salts for example?
Where did you get those programs if I may ask?
[Edited on 28-7-2018 by nitro-genes]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Simply red,
There is a popular equation to product density which only require 2 numbers, the mass of the compound, and space it takes up. However, here we have no
crystal structure, so this equation is rather unaccurate, it has an average error of 0.09g/cm3.
The equation I used is based on the same idea, however I take into account the electrostatic interactions of a compound, this pulls the average error
from 0.09g/cm3 down to 0.03g/cm3.
https://www.tandfonline.com/doi/abs/10.1080/0026897100370222...
Nitrogenes: Yes, the basis set is used as stated for the equation, since the equation (from above link) states to use B3PW91, then I use B3PW91, since
the numbers in the equation is optimized for the basis set. The other research team accidentally used B3LYP. They do produce similar results, however
you should be checking twice about really publishing a data of a CHON compound that exceeds 2.1g/cm3.
The software are mostly free, you can ask to obtain a list of a variety of software such as ORCA, MOPAC, GAMESS, multiwfn. They're rather hard to set
up on windows.
>Does it also work for salts for example?
Yes there is an equation for salts.
>how does it estimate the influence of between molecule interactions?
trust me, im just calculating for the results to plug into the equation, quantum mechanics is out of my reach 
[Edited on 28-7-2018 by DubaiAmateurRocketry]
|
|
|
simply RED
Hazard to Others
Posts: 209
Registered: 18-8-2005
Location: noitacoL
Member Is Offline
Mood: booM
|
|
>Does it also work for salts for example?
Yes, if you have the crystal structure and optimize a periodic cell (with VASP, CP2K, other software). With a single ion pair, no way.
>how does it estimate the influence of between molecule interactions?
It does not (with a single molecule), though for substances that have only electronegative, like ONC (or positive) centers around the molecule, the
error may be significant. If it has alternating positive - negative centers, the error may be negligible.
How to guess a crystal structure of a new molecule? Absolutely impossible for now (to get the real structure), with any supercomputer or software.
Also, you optimize it with B3LYP anyway, why calculate the heat of formation with PM3? Just do all calculations with one method and one basis set.
[Edited on 29-7-2018 by simply RED]
When logic and proportion have fallen sloppy dead...
|
|
|
Rocinante
Hazard to Others
Posts: 121
Registered: 13-11-2017
Member Is Offline
Mood: No Mood
|
|
Could you investigate HMTD? There are conflicting reports on it. Also, 2,4,6-trinito-1,3,5-triazine would be nice.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Why is it so difficult to determine crystal structure? It seems as if the process of crystallization is very similar to in silico protein folding,
where a monomer subunit is added one at the time and a "local folding minimum" is calculated IIRC, before adding a next monomer subunit and repeating
the process. It seems as this would be the most natural approach to the problem, not unlike happens in reality. So why is this not feasible? No
covalent bond like for proteins probably means a lot more possibilities to calculate, but maybe some assumptions can be made here.
[Edited on 29-7-2018 by nitro-genes]
|
|
|
simply RED
Hazard to Others
Posts: 209
Registered: 18-8-2005
Location: noitacoL
Member Is Offline
Mood: booM
|
|
Too many possible crystal structures with similar energies. And when using classical mechanics, you can not pick up the natural right one. Using QM is
too expensive as crystal structures are large. Anyway, an assumption can be made, that will not be quite far from reality, at least in energy and
density.
Proteins have more key-keyhole motives and are maybe slightly easier to "fold" than crystals to "crystallize". Also, to point out the obvious, all
classical methods are parametrized, made to work well, for proteins/DNA/RNA.
In my experience maybe 80% of docking and protein folding was incorrect, if not combined with crystallography in some way. So, it is extremely
difficult with biomolecules too.
The correct approach is the combination of computational methods and synthesis and crystallography/IR/NMR. Filling the blanks of one method with the
other.
When logic and proportion have fallen sloppy dead...
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Quote: Originally posted by Rocinante  | | Could you investigate HMTD? There are conflicting reports on it. Also, 2,4,6-trinito-1,3,5-triazine would be nice. |
There is already a lot of papers on trinitrotriazine, it cannot be synthesized, and therefore has no value in predicting its detonation properties.
HMTD information is widely available.
|
|
|
Rocinante
Hazard to Others
Posts: 121
Registered: 13-11-2017
Member Is Offline
Mood: No Mood
|
|
yeah, both both have differing values in the literature.. by about 15 % or more
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Simply red, thanks for the great reply! The existence of many different crystal polymorphisms seems to exclude a "single crystal lattice" anyway
indeed...
@ DubaiAmateurRocketry:
This one seems theoretically possible (Couldn't access paper, no idea at what scale/yield these dimers can be synthesized):
Solid state [2 + 2] photocycloaddition for constructing dimers of N,N′-diacyl-1,4-dihydropyrazines based on thiourea-induced assembly
Qiangwen Fan, Xiaowei Duan and Hong Yan* (2018)

[Edited on 30-7-2018 by nitro-genes]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Red "why calculate the heat of formation with PM3? Just do all calculations with one method and one basis set. "
PM3 or PM6 give direct HOF output, DFT based HOF is extremely hard to calculate.
Nitro-genes,
I will give that compound a run later tonight, it indeed seems extremely interesting. By itself, its a bit too oxygen-negative, but the structure
itself and the potential for functionalization is interesting.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Hats off for being able to calculate these properties almost from scratch! Would like to give it a go myself, maybe starting with just density. What
is an easy way for calculating density? Not wanting to sound lazy, is there a freeware program that can make reasonable estimates of density from the
unit cell volume/molar mass method directly from a structural 3D drawing?
Compared to HNIW it would be rather oxygen deficient indeed, not sure if the dimerization process would tolerate other functional groups present, if
so more oxygen balanced and dense compounds maybe could be derived from it. It seemed to have the potential for being very dense, the 3D structure
seemingly adopts an almost cubane like structure with the nitramine groups protruding from the planes. Not sure if determining density by eyeballing
it is very accurate though.  Are there some general rules for example why some
compounds are much more dense than others? For example, why is the 5-6-5 configuration so dense, it doesn't seem directly apparent by looking at it's
structure/volume? Are there some general rules for example why some
compounds are much more dense than others? For example, why is the 5-6-5 configuration so dense, it doesn't seem directly apparent by looking at it's
structure/volume?
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
nitrogenes:
So far, for heat of formation.
PM6 gave a 195 kJ/mol.
PM3 gave a 361 kJ/mol.
the skeleton itself has 155.1 kJ/mol, a little bit less bond strain than I imagined, but pretty decent.
cubane skeleton has a calculated HOF of around 470 kJ/mol using PM3.
This is expected, as while NNO2 and the cube structure is positive, the 8 CH is rather not contributing at all. This compound needs functionalization
on these CHs, it would become an insanely high energy high density compound if we hanged NO2 groups off the carbons too.
Usually PM6 is a little more accurate. This structure is extremely symmetric, functionalizing it might produce some limit breaking HEDMs.
I have not calculated the density yet.
I uploaded a paper for you nitro-genes, for group-additivity for density. The hand calculations is a bit difficult, however faster than a computer
currently, but also, somewhat less accurate.
the time to optimize a structure scale with the number of nitro groups.
HHTDN took my high-end 2016 PC over 30 hours to optimize. Yup, you need like 30 or 40+ cores to run a bunch of stuff at once or at a faster speed,
basically a super-computer.
For eye-balling a compounds density, symmetry, groups contributions, may a factor. Usually NO2 is highly favorable for density, NH2 is decent if nitro
or N-oxides exist, azido group is okay, usually not very dense. N-oxides could be highly dense, or not if its the sole functional group in a
heterocyclic compound (ONC and TTTO's oxygen surrounded structure caused repelling intermolecular forces that reduced its density).
Attachment: Kotomin and Kozlov 2005.pdf (90kB)
This file has been downloaded 466 times
[Edited on 31-7-2018 by DubaiAmateurRocketry]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Nitro genes,
your calculations have started, expect at least 12 hours
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
12 hours computing time still beats 12 minutes of calculating by hand IMO. Accurate calculation of EM properties by computational methods is a great
tool, I think this thread should be stickied right away. Some more information on the "methods" and programs used, with some illustrated calculated
examples for example would be great as well
155.1 kJ/mol, that is pretty decent indeed, somewhat more than HNIW IIRC. Really curious what the density will turn out to be. Not sure if adding more
NO2 groups to the carbons would be possible or stable. Talking about increasing oxygen balance....would a putative dimer of RDX exist as well? Wondered why ONC and TTTO were less dense than expected, thanks for pointing that
out! That paper you mentioned about the limits possible for CHNO explosives seems like an interesting read as well.
[Edited on 31-7-2018 by nitro-genes]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
see U2U
|
|
|
simply RED
Hazard to Others
Posts: 209
Registered: 18-8-2005
Location: noitacoL
Member Is Offline
Mood: booM
|
|
You can directly calculate heat of detonation, formation, whatever with DFT or any other method.
dH (molecule) - dH(products).
If possible dH has to be ZPE corrected (zero-point energy). If not, it is not a big mistake (some kJ/mol).
When logic and proportion have fallen sloppy dead...
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Another thing I was wondering (excuse the spoonfeeding request):
Sometimes when drawing a particular structure of an EM I wonder if (apart from being able to synthesize it in practice) would be able to exist at all
under normal conditions. How does one calculate whether a putative EM exist at room temperature or only at absolute zero for example?
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
| Quote: Originally posted by nitro-genes | Another thing I was wondering (excuse the spoonfeeding request):
Sometimes when drawing a particular structure of an EM I wonder if (apart from being able to synthesize it in practice) would be able to exist at all
under normal conditions. How does one calculate whether a putative EM exist at room temperature or only at absolute zero for example?
|
I could post some methods soon on the heat of formation and density, but I'm currently calibrating and comparing some of these methods so I can post
the easiest and most efficient method here.
regarding your question, it'd be reasonable to design theoretical compounds based on existent synthesis and precursor. Also, we would not need to know
if it'd be stable at 0K, since there would be almost zero application for an EM if it is not ambient-stable. Pentazoles are room-temp-stable, however
cg-polynitrogen is only stable at pressures higher than what is needed to synthesize diamonds.
As for actually calculating these properties, that is rather complex.
|
|
|
simply RED
Hazard to Others
Posts: 209
Registered: 18-8-2005
Location: noitacoL
Member Is Offline
Mood: booM
|
|
All methods discussed so far deal with geometry optimization of the molecules. As it is known, the geometry optimization gives properties at 0 K or
very close to it. So basically, if you try optimizing a molecule, it has to be very unstable for the optimization not to converge to the initial
geometry.
If you want to do calculations at T > 0 K, try quantum dynamics. For example with CP2K. My PhD thesis was about this type of calculations, but on
bio molecules and not EMs. I've tried EMs and it works too.
I only used the software, did not write it.
https://www.cp2k.org/
[Edited on 12-8-2018 by simply RED]
When logic and proportion have fallen sloppy dead...
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
| Quote: Originally posted by simply RED | All methods discussed so far deal with geometry optimization of the molecules. As it is known, the geometry optimization gives properties at 0 K or
very close to it. So basically, if you try optimizing a molecule, it has to be very unstable for the optimization not to converge to the initial
geometry.
If you want to do calculations at T > 0 K, try quantum dynamics. For example with CP2K. My PhD thesis was about this type of calculations, but on
bio molecules and not EMs. I've tried EMs and it works too.
I only used the software, did not write it.
https://www.cp2k.org/
[Edited on 12-8-2018 by simply RED] |
theres a lot of methods where you could convert 0K to 287k, some CBS methods give less than 3kcal deviation, others give less than 1kcal, however, are
more computationally stressful. Some really accurate methods lack published data and their accuracy on energetic materials.
for amateurs, PM7 is enough, it gives ~10-15 kcal deviation.
[Edited on 12-8-2018 by DubaiAmateurRocketry]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
I am running into little troubles while using CBS-4M and B3LYP based HOF calculations, occasionally, some compounds produce a surprisingly wrong
result. For now, I will post a PM7 vs PM6 vs PM3 result.
PM7 is the most efficient method at calculating HOF. Although there is still a high deviation on some molecules, it is probably a little bit better
than a guesstimation.
PM7 can be accessed with the free software MOPAC, which could be found using google, just follow closely to its installation guidelines.
PM6 and PM3 can be found in many softwares, Gaussian, MOPAC, etc.
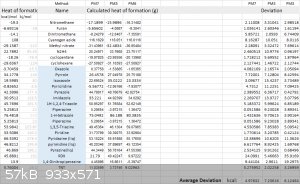
[Edited on 16-8-2018 by DubaiAmateurRocketry]
[Edited on 16-8-2018 by DubaiAmateurRocketry]
|
|
|
| Pages:
1
2 |










