| Pages:
1
2
3 |
Bottle
Harmless

Posts: 8
Registered: 15-2-2018
Member Is Offline
Mood: No Mood
|
|
Reduction of substituted 2-phenylnitroethenes to 2-phenylnitroethanes with NaBH4 - Maybe the greatest way to make tar
Hi!
I need some input
I'm currently trying to make 2,5-dimethoxy-phenylnitroethane from the 2,5-dimethoxyphenylnitroethene (2,5DMNS) counterpart
I tried several systems found on the net:
| Quote: |
In a two-neck 250ml rb flask with a egg shaped stirbar containing 40ml EtOAc and 10ml denaturated EtOH(1), 3.63g (95mmol) commercial NaBH4 is
added in one portion. The flask is immersed in a 10°C cooling bath. A 4-bulb Allihn condenser(2), with cold tap water running through it, is inserted
in the central neck and a thermometer in the side neck. Let the temperature drop to 15°C and start add 5g (24mmol) 2,5-dimethoxy-beta-nitrostyrene in
0.5g portions to the borohydride suspension. Keep the temperature between 20-30°C during the addition, which takes some 15 minutes. When all
nitrostyrene has been added allow the reaction mixture stir for another 20 minutes.Add 50ml cold water to the reaction mixture and stir for a couple
of minutes. Transfer the mixture to a 500ml separation funnel and remove the bottom aquoeous layer(3). Add another 50ml portion of cold water, shake,
allow to separate and remove bottom layer. Add a third portion of 25ml cold water and dropwise 50% aq. acetic acid untill gas evolution ceases. One
might need to shake it now and then to bring the aqueous layer in contact with some borohydride still remaining on the walls. When no more gas is
evolved add 50ml brine, shake, let separate and remove the bottom layer. Now you will have a bright yellow solution of the phenylnitroalkane in some
wet EtOAc, dry with some MgSO4 and strip off the solvent to give a yellow oil. Yield 4.94g (98%) 1-(2,5-dimethoxyphenyl)-2-nitroethane, purity 97%
(HPLC). |
Source: https://erowid.org/archive/rhodium/chemistry/nitrostyrene.na...
I perfomed it in a 400mL beaker instead of a flask, except that I followed the instructions very carefully, but ending up with a slightly dark reddish
oil at the end.
Shortpath-destillation in good aspirator vacuum and a 130°C oil bath yielded within a few minutes only a miniscule amount of condensate (a few mgs)
of a colorless imho pleasent sligthly nitromethane- and desifectant like smelling colorless oil in the collecting flask and a violently fast
decomposing of the oi which began to turn into sticky black tar within a very few minutes so destillation was discontinued
| Quote: |
10,5 g (50 mmol) 2,5-dimethoxy-beta-nitrostyrene was added during 5 minutes to a solution of 2,4 g (63 mmol, 1,25 mol eq.) sodium borohydride in
50 ml IPA and 20 ml water. The temperature rose from 20 to 50°C while the orange color faded to a slightly yellow. 2N HCl was then carefully2 added
until pH 4 was reached followed by enough solid NaCl to cause the IPA to form a separate layer containing the product. Two volumes of water was added
to the isolated IPA layer which caused 2-nitro-1-(2,5-dimethoxyphenyl)ethane to separate as a clear yellow oil and was isolated by extraction with
2x15 ml toluene. The organic phase dried over MgSO4 and the solvent removed under vacuum leaving the product as a clear yellow oil. Yield 9,7 g (46
mmol, 92%) 2-nitro-1-(2,5-dimethoxyphenyl)ethane. |
Mirror: https://chemistry.mdma.ch/hiveboard/novel/000426052.html
I used ~30% HOAc instead of HCl to bring the pH to 4.00 and used a 300ml erlenmeyerflask with ice-bath-cooling instead of RT to avoid dimeric
sideproducts (maybe a mistake?)
In my case the isolated sligthly yellow IPA-layer precipitated around 1g of a grey-greenish very fine solid in the IPA layer while washing it the
second time with sat. aq. NaCl-solution. I filtered and washed the cake with cold IPA and removed some IPA using mild heat from waterbath and an
aspirator pump. I wanted to see if even more will precipitate when cooling but upon cooling in the fridge to my suprise another sideproduct was formed
- 1-2mL of a very viscous deep orange-red oil which is heavier than brine crashed out, causing a beautyful 3-layer system in the s.funnel with
a clear bright yellow upper layer, a clear colorloess brine center and a clear deep orange red bottom-layer
Not gone any further yet, don't know if it's worth a try
I used 99.98% IPA, home-destilled water, high quality 99% anhydrous EtOH with 1% MEK, 99.8% EtOAc, food-grade glacial acetic acid, technical
borohydride. The nitrostyrene was made with catalytic amount of EDDA in hot IPA and then recristallized twice from EtOAc which caused
up to 2cm long needles which had to be grinded for further use. Except the technical grade borohydride all chemicals were from exceptional quality, so
I doubt that would be the problem.
Now a few questions?
-What are these impurities I was able to isolate?
-What causes them, especially what causes tar when heating slightly? Air? Impurities in technical borohydride? Does EDDA interfer even in trace
amounts? Reactions with the solvent?
Has anyone any clue about that?
-Might I have sepperated ALL the impurities due their insoloubility in IPA?
-Has anyone here had success or know someone personally who had success to produce a acceptable yield of a product of a usable quality?
-I can't find a literature boiling point of the desired product, is a good aspirator vacuum-pump good enough to destill it if it's pure?
The impurity seems to have almost catalytic-self-destruction impact on the substrate, the oil I recieved from the first procedure produces tar already
at very moderate temperatures around 50-70°C.
(Side-Question: Is there a way to get the aldoxime from the NS without the usage of exotic homogenous Rhodium- or Gold/TiO2-Catalysts or tons of acid
in acceptable yield (I have Pd/C!)? Would be a nice bypass!? I know it's possible with longer chained phenylnitroalkanes, but is it possible with
phenylnitroethanes aswell?
[Edited on 15-2-2018 by Bottle]
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Online
Mood: Quarantined
|
|
Do you intend to make 2C-H ( 2,5-DIMETHOXYPHENETHYLAMINE) as your final goal?
Pihkal say that's a no psychoative amine at all.
If so, read the paper attached about reduction of nitrostyrenes with Zn and HCl at 0ºC.
I hope it helps.
Attachment: phpmeHwOU.pdf (1.1MB)
This file has been downloaded 1267 times
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
First try? Don't improvise. Follow instructions exactly. If they say HCl, use HCl.
Hard enough to get good results, when conditions are followed exactly.
Just because someone said the procedure works, doesn't mean it does.
|
|
|
Bottle
Harmless
Posts: 8
Registered: 15-2-2018
Member Is Offline
Mood: No Mood
|
|
That paper looks rather unsexy to me, as workup is hard.
It's not my first try, it's actually already my third try using this substrate. I've done it on other substrates with success, though this one is a
bitch.
I used acetic acid instead of HCl to avoid Nef-reaction side products as I only got dirt on the very first try when using HCl and I guess it might by
caused by Nef-reaction (substrate seems very sensitive towards this). Acidfication only has to be made to release product from their nitronate salts
and thus a weak acid can be used for sure, avoiding Nef-reaction
I made a TLC (PE:EToAC: DCM:Ethylenediame) on the yellow IPA layer, it shows one big spot along with a tiny one (hopefully no the regarded product)
and no traces of the educt.
Evaporation of the IPA yielded only 30% of theory of a yellow-to-orange oil
CTH Reduction with Ra-Ni+Formic-Acid (5mmol substrate;3mL formic, 3mL EtOH and one spatula of Ra-Ni heated to ~50°C similar to this ref: http://eprints.uni-mysore.ac.in/4673/1/00397910008087439.pdf) is currently running - at least some kind of reaction is taking place, as CO2
bubbles are beeing formed
Though I know that 2C-H and educt almost have the same retention times for eluents like mine, so TLC will be hard on this one
[Edited on 20-2-2018 by Bottle]
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
Acetic acid should work too, I've seen this procedure use both. I don't think that's the problem.
How soluble is the nitrostyrene in isopropanol? What was going on at the bottom of the flask? How finely divided was your nitrostyrene?
Come to think of it, did you even have any nitrostyrene? Are you sure you didn't just recrystallize your benzaldehyde? I have to ask dumb questions
too, after all. I'm assuming you used this procedure?
https://erowid.org/archive/rhodium/chemistry/edda.html
Was it a vivid orange color? Were there a lot of bubbles when you added the nitrostyrene? Did you have stirring?
Also, if your sodium borohydride is exposed to air for a long time, it'll turn into borax. However if that were the case there would be no reaction.
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
Bottle
Harmless
Posts: 8
Registered: 15-2-2018
Member Is Offline
Mood: No Mood
|
|
In the 1g batch of inpure phenylnitroethan (from IPA-route) I got only about 230mg 2C-H-base-carbamate (tested via TLC) with the Ra-Ni/Formic method
and a lot of side products (tar!) making workup horrible so I'm still not happy with the quality of the phenylnitroethane
Quote: Originally posted by Melgar  |
How soluble is the nitrostyrene in isopropanol? What was going on at the bottom of the flask? How finely divided was your nitrostyrene?
|
The NS is only sparingly soluoble in IPA
The NS was not very finley divided prior usage (maybe the problem?)
| Quote: |
Come to think of it, did you even have any nitrostyrene? Are you sure you didn't just recrystallize your benzaldehyde? I have to ask dumb questions
too, after all. I'm assuming you used this procedure? Was it a vivid orange color? Were there a lot of bubbles when you added the nitrostyrene? Did
you have stirring?
|
NS was recristallised twice and it fits literature melting points and is clean by TLC, no doubt it's the correct stuff. It has a very vivid deep
orange color. There were only tiny bubbles while adding the NS. I had very strong magnetic stirring.
| Quote: |
Also, if your sodium borohydride is exposed to air for a long time, it'll turn into borax. However if that were the case there would be no reaction.
|
Yes my borohydride hasn't been used since a few years and might have lost a lot of quality. I will get some new stuff
[Edited on 28-2-2018 by Bottle]
|
|
|
cubalibre
Harmless
Posts: 11
Registered: 3-10-2016
Member Is Offline
Mood: H2 saturated
|
|
On addition the reduction is normally vigorous and very exothermic which is the only reason this NS goes into solution in that IPA/H2O compared to
EtOAc/EtOH system.
You will see a fast color change from orange to white/clear/cream color on addition of the substrate.
Did you notice this and H2 production on quenching with GAA?
[Edited on 2-3-2018 by cubalibre]
|
|
|
clearly_not_atara
International Hazard
Posts: 2788
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
The nitrostyrene precursor reacts with the nitroalkane product in a Michael addition. So if the reduction is incomplete you will get a dimer upon
distillation. That is the main pitfall of this procedure. So if your borohydride is bad it can muck up the whole thing.
Maybe a nitroalkene scavenger could be used if you think borohydride is too expensive to use in excess. I'm not sure what would be suitable; perhaps
sodium phenolate would work.
Alternatively the Zn/HCl reduction suggested by Chemi Pharma is preferred for this substrate. I highly recommend it if you want to make a
phenethylamine.
|
|
|
Corrosive Joeseph
National Hazard
Posts: 915
Registered: 17-5-2015
Location: The Other Place
Member Is Offline
Mood: Cyclic
|
|
I have previously uploaded an excellent review on the reduction of nitrostyrene using the Zn/HCl system in the link below......
'Optimizing Henry reaction conditions (substituted benzaldehydes + nitromethane)'
https://www.sciencemadness.org/whisper/viewthread.php?tid=76...
/CJ
Being well adjusted to a sick society is no measure of one's mental health
|
|
|
Bottle
Harmless
Posts: 8
Registered: 15-2-2018
Member Is Offline
Mood: No Mood
|
|
I think I have found the reason(s)
--->The nitrostyrene was rextallised a fourth(!) time, but this from EtOH/HOAc 1:1 using a lot active charcoal and filtration, which didn't improve
the melting point but did remove a lot of orange color, which made the nitrostyrene brighter orange, almost yellow! Using this system also produces
less loss that rextallisation from EtOAc<---
I guess the EDDA-way-made nitrostyrene is directly only usable for LAH reduction without heavy cleaning.
Then I used 5g nitrostyren to 4g freshly bought NaBH4, dissolved the NS in a lot (~100mL) EtOAc and added it over 2h drop by drop to the fresh NaBH4
suspended in 40mL ahydrous EtOH in a 200mL beaker. I don't know the yield as I had an overboiling during acidification (next time I would use
ice-cubes), though this time the product seems MUCH cleaner, as it had a different less pleasent smell and almost no tar formed 
I will post again if it's still not pure enough for decent yielding CTH reduction with Formic/Raney etc.
Thx guys, especially to Melgar!
[Edited on 11-3-2018 by Bottle]
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Online
Mood: Quarantined
|
|
Man, I could tell you a better way to use your NaBH4 in addition to NiCl2 to perform your synthesis withouth any tar formation and with high yields,
but you are a newbie here and came, at your first time, just asking how to perform a nitroalkene reduction drugs related.
Don't you think it's a little suspect? A lot of other members read this thread but nobody until now, with a few exceptions, didn't say a word, cause I
suspect they feel this thread is very suspicious too, I guess!
Search across Sciencemadness about my nickname and I'm sure you will find threads about NaBH4 couple that will help you, certainly, with your
synthesis.
It's not tolerated by moderators spoonfeeding here at the forum, neither I want to do it so for you.
|
|
|
FireLion3
Hazard to Others
Posts: 218
Registered: 11-1-2014
Member Is Offline
Mood: No Mood
|
|
I have to agree with Chemi Pharma. While discussing the theory and synthesis of restricted compounds isn't totally frowned upon on this forum, doing
so for simple cookery is. It would be a totally different matter if OP was a genuine researcher exploring subtle chemical dynamics for
academic/research or whatever, but this isn't the case. It's pretty clear OP is a beginner attempting to make a psychoactive drug (one that is illegal
in most countries), who hasn't been studying chemistry for very long - no offense OP. Especially using words like "rextallised" and "rextallisation"
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
Eh, it was borderline, and fairly benign. We don't even know where he is to know if he's breaking the law. And he seemed to be more interested in
the chemistry than how fucked up he's going to get, so that's a point in his favor. As long as he learned something about chemistry, and as long as
he's not going to just forget it as soon as he's done with this reaction, I have no problem with asking and answering these types of chemistry
questions.
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
Corrosive Joeseph
National Hazard
Posts: 915
Registered: 17-5-2015
Location: The Other Place
Member Is Offline
Mood: Cyclic
|
|
I don't see a problem here
I am much impressed by the characterization and the tenacity to keep going despite alternative suggestions by members
This is far above the many shitty cookery posts by first time members.
Target compound isn't even active
/CJ
[EDIT 2] - I won't post again, so I'll drop it here.
Can we stop spamming an otherwise decent thread......?
[Edited on 15-3-2018 by Corrosive Joeseph]
[Edited on 15-3-2018 by Corrosive Joeseph]
Being well adjusted to a sick society is no measure of one's mental health
|
|
|
Vosoryx
Hazard to Others
Posts: 282
Registered: 18-6-2017
Location: British Columbia, Canada
Member Is Offline
Mood: Serial Apple Enjoyer
|
|
It isn't active, so I think this is allowed.
"I was up to a 1+ with 80 milligrams of the hydrochloride, and since it appeared to be totally a physical trip with tremors and some cardiovascular
push and nothing of a sensory nature, I chose to explore it no further." - Alexander Shulgin (PiHKAL)
Edit:
My reply was very similar to CJ's... which I only saw after posting. I'm not copying you, I swear!
[Edited on 15-3-2018 by Vosoryx]
"Open your mind son, before someone opens it for you." - Dr. Walter Bishop
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Online
Mood: Quarantined
|
|
I don't know, I'm still felling something smelling bad here.
Don't forget guys 2C-H is easy brominated to afford 2C-B, a banished, psychedelic and aphrodisiac drug, with efects similar to Exctasy (MDMA).
See what Pihkal say about the easy bromination:
"To a well-stirred solution of 24.8 g 2,5-dimethoxyphenethylamine in 40 mL glacial acetic acid, there was added 22 g elemental bromine dissolved in 40
mL acetic acid. After a couple of min there was the formation of solids and the simultaneous evolution of considerable heat. The reaction mixture was
allowed to return to room temperature, filtered, and the olids washed sparingly with cold acetic acid. This was the hydrobromide salt. "
Even 12-24 mg is enough for an efetive dosage and to fell its effects.
|
|
|
Bottle
Harmless
Posts: 8
Registered: 15-2-2018
Member Is Offline
Mood: No Mood
|
|
Neither 2C-H or my target are illegal where I live.
It's not against the forum-rules.
If we start talking like "but you could easily do xxx with it" we could directly ban all kind of chemistry and chemistry-related knowledge! 
Back to topic
Experiment still was unsuccessful.
Everything I got from the last try (check my last post) was an oil which was much denser than water but didn't reduce to the desired product with
Pd/C+H2@10bar until hydrogen uptake ceased plus some additional hource. A/B-Extraction after H2/Pd/C yielded only a lot of a viscous heavy oil in the
from the washes of the acidic media and a tiny amount of some alkaline plastic-like soft but not liquid material (maybe the reduced dimer), the
desired product was only visible by TLC. My latest guess is that some styrene-->polystyrene-alike oligomerisation takes place when the solvent is
evaporated, maybe catalysed by an inpurity.
Original sources used a rotovap, while I was only using a hot water bath and water-jet-vacuum.
Though anyway there must be much more going on than only Micheal-addition to a dimer, as almost zero yield is produced which can't be explained
easily. Any ideas?
I might try it again without solvent removal, using the intermediate in solution as-is. Or Maybe with dioxane/silica like in the original sources. Is
a acidic workup / neutralisation of the intermediate necessary as with hydroboration/oxymercuration etc.? I can't find enough information about the
mechanism.
@ChemiPharma
| Quote: | | Man, I could tell you a better way to use your NaBH4 in addition to NiCl2 to perform your synthesis withouth any tar formation and with high yields,
but you are a newbie here and came, at your first time... |
I was already thinking of using NaBH4 or KBH4+Raney-Nickel (as potassium borohydride is a better hydrogen donor for CTH
as far as I know http://www.arkat-usa.org/get-file/23388.html) as one-pot reaction which should be almost exactly the same way as your NaBH4+NiCl2 which produces
nano-nickel on nickel-boride in-situ as active catalyst, making it a very similar CTH-mechanism for the nitro-reduction aswell. Though I don't know if
there might be any side-reaction as there might be some kind of a hydroborate-complex forming with the double-bond, especially under alkaline
conditions, which might react to a saturated nitroalcohol(-->aminoalcohol) and more complex sideproducts.
So, have you ever tried it (maybe with a similar substrate) or why are you telling that there's s high yield and no tar produced?
Can anyone here explain the mechanism how the nitro- activated (conjugated) double-bond is actually reduced? Is it more 1,4-addition-alike funky or
more hydroboration-alike or something completly different?
[Edited on 15-3-2018 by Bottle]
|
|
|
clearly_not_atara
International Hazard
Posts: 2788
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
We're not the police, it's hardly our job to speculate about what people
might be doing. We had a sticky on 2,5-dimethoxybenzaldehyde at one time and everyone knows what that's for (not to mention the crucial role
of acetic anhydride in making heroin!). Our concern is with what they tell us they're doing and, more importantly, with what we're doing,
which is to spread the understanding of chemistry, but not to promote or in any way encourage drug manufacturing. That chemistry makes drug
manufacturing possible is hardly something we can hope to avoid; in fact the chemical restrictions we now fight so hard against came from the idea
that the government could ban only drug chemistry, which as we all know turned into a ban on all chemistry.
Frankly I think it's worse to post quotes and links from PiHKAL than to answer questions about reducing nitrostyrenes. We don't need to give him any
more help making drugs; if someone posts repeated queries about each step in the 2C-B manufacturing process, we can cross that bridge when we come to
it, and ignore or ban them with good justification.
Also, 2C-B may be active, but it was hardly popular last I checked, which suggests that at least OP is not going to make any money -- and commercial
production is the real problem, anyway.
[Edited on 15-3-2018 by clearly_not_atara]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
2C-H is a precursor to other designer phenethylamines and these days is a controlled substance like lysergic acid.
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Online
Mood: Quarantined
|
|
Since some members here think that's nothing wrong, we're not the police and forum's moderators did nothing about this thread until now, I feel free
to expose my ideas and reply @bottle question, although, in my opinion, I still thinking this discussion obviously are drugs related:
| Quote: Originally posted by Bottle | @ChemiPharma
| Quote: | | Man, I could tell you a better way to use your NaBH4 in addition to NiCl2 to perform your synthesis withouth any tar formation and with high yields,
but you are a newbie here and came, at your first time... |
I was already thinking of using NaBH4 or KBH4+Raney-Nickel (as potassium borohydride is a better hydrogen donor for CTH
as far as I know http://www.arkat-usa.org/get-file/23388.html) as one-pot reaction which should be almost exactly the same way as your NaBH4+NiCl2 which produces
nano-nickel on nickel-boride in-situ as active catalyst, making it a very similar CTH-mechanism for the nitro-reduction aswell. Though I don't know if
there might be any side-reaction as there might be some kind of a hydroborate-complex forming with the double-bond, especially under alkaline
conditions, which might react to a saturated nitroalcohol(-->aminoalcohol) and more complex sideproducts.
So, have you ever tried it (maybe with a similar substrate) or why are you telling that there's s high yield and no tar produced?
|
Yes I did nitroalkenes reduction reaction using NaBH4 and NiCl2 in methanol a few times, just for cientific purposes. And I'm the major defensor of
this technique of reduction here at the forum, instead using expensive Pd/C, Raney nickel and surpassed techniques like urushibara nickel, Al/Ni+2 and
Hg/Al amalgam, that some members here still defending.
You can follow the directions given by the papers I attached below to have high yields getting your 2C-H with no tar at all. Remember tar is
intimately related with room temperatures reaction mix and above. However the papers didn't say a word about temperature factor, try to do the
reactions at near 0ºC:
Attachment: sodium borohydride-nicl2-reduction of nitro compounds.pdf (399kB)
This file has been downloaded 1247 times
Attachment: amphetamine synthesis from phenylnitropropene - NaBH4 + NICL2.doc.doc (189kB)
This file has been downloaded 1884 times
[Edited on 15-3-2018 by Chemi Pharma]
|
|
|
Corrosive Joeseph
National Hazard
Posts: 915
Registered: 17-5-2015
Location: The Other Place
Member Is Offline
Mood: Cyclic
|
|
| Quote: Originally posted by Bottle |
Can anyone here explain the mechanism how the nitro- activated (conjugated) double-bond is actually reduced?
|
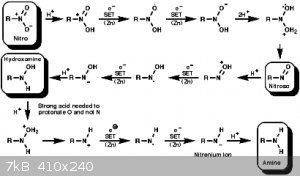
Check Hudlicky's for an excellent review on nitro and nitro conjugated double bond reductions and mechanisms.
Maybe you know it already...... Pages 69 thru to 75
The whole book really is a must-read anyway.
'Reductions in Organic Chemistry' - Milos Hudlicky
http://cnqzu.com/library/Anarchy%20Folder/Chemistry/Crystali...
Also attaching Nitro to Amine SET mechanism - taken from
http://www.ch.ic.ac.uk/widdowson/teach_files/nitrogen/dw1.ht...
And you might like this -
'Catalytic Transfer Hydrogenation of Nitroalkenes to Primary Amine'
Attached -
It has a proposed hydride transfer mechanism on beta-Nitrostyrene deep in there somewhere.
And this for a chaser -
'Selective Reduction of Conjugated NitroOlefins' - https://etd.ohiolink.edu/!etd.send_file?accession=osu1448465...
I'm sure I have more but that'll do for now.....
Very interested in your Ni/Formic Acid. I sometimes wonder if a Nickel nanoparticle CTH could replace Ra-Ni which is not so OTC but your results with
that don't look so promising.
Hope this helps
/CJ
Attachment: Catalytic Transfer Hydrogenation of Nitroalkenes to Primary Amine.pdf (1.4MB)
This file has been downloaded 943 times
Being well adjusted to a sick society is no measure of one's mental health
|
|
|
Bottle
Harmless
Posts: 8
Registered: 15-2-2018
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Chemi Pharma |
Yes I did nitroalkenes reduction reaction using NaBH4 and NiCl2 in methanol a few times, just for cientific purposes. And I'm the major defensor of
this technique of reduction here at the forum, instead using expensive Pd/C, Raney nickel and surpassed techniques like urushibara nickel, Al/Ni+2 and
Hg/Al amalgam, that some members here still defending.
You can follow the directions given by the papers I attached below to have high yields getting your 2C-H with no tar at all. Remember tar is
intimately related with room temperatures reaction mix and above. However the papers didn't say a word about temperature factor, try to do the
reactions at near 0ºC:
[Edited on 15-3-2018 by Chemi Pharma] |
Thank you very much!
But have you also tried it on nitrostyrenes or only on a-methyl-nitrostyrenes? They behave quite different; a-alkyllnitrostyrenes are generally very
easy to reduce with NaBH4 to their nitroalkanes while the nitroethenes tend to produces crap!
[Edited on 16-3-2018 by Bottle]
|
|
|
Bottle
Harmless
Posts: 8
Registered: 15-2-2018
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Corrosive Joeseph |
...
Very interested in your Ni/Formic Acid. I sometimes wonder if a Nickel nanoparticle CTH could replace Ra-Ni which is not so OTC but your results with
that don't look so promising.
|
I've used Ra-Ni/Formic a few times times on completly different substrates for nitro reductions in EtOH, usually always got good (70-90%) yields, the
30% yield is most likes caused by very impure educt.
It's imho as good as Pd/C+Formate in MeOH, maybe a bit smoother and workup is sometimes crappy as a little Ni can dissolve (though not so much when
using formate instead of formic acid).
I bet CTH will work with NiNP and nickel boride aswell.
Most likely even with Ni2+ and heat, producing nanoparticles in-situ, though of course with limited usage
edit j_sum1
fixed formatting issue
[Edited on 17-3-2018 by j_sum1]
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Online
Mood: Quarantined
|
|
| Quote: Originally posted by Bottle | Thank you very much!
But have you also tried it on nitrostyrenes or only on a-methyl-nitrostyrenes? They behave quite different; a-alkyllnitrostyrenes are generally very
easy to reduce with NaBH4 to their nitroalkanes while the nitroethenes tend to produces crap! |
To reduce methyl nytrostyrenes and nitropropenes you'd better use nickel boride instead NaBH4/CH3OH couple alone, while reducing nitrostyrenes itself,
I'd rather use Zn and HCl at 0ºC, as I've told before in this thread.
Don't make sense to me reducing nitroalkenes to nitroalkanes intermediates if the target are the amines and I can do it directly, in just one step.
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
I'm posting here just to fix the formatting glitch that XMB seems to be having right now.
edit: Bottle, or a moderator: please edit that post, and replace the curly bracket with a square bracket for the rquote tag, ok?
[Edited on 3/16/18 by Melgar]
[Edit j_sum1]
Thanks for that Melgar.
Done.
[Edited on 17-3-2018 by j_sum1]
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
| Pages:
1
2
3 |









