semiconductive
Hazard to Others

Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Making a micro-buchner vacuum filtration wand.
I've been doing analytic chemistry, and trying to determine accurate masses of precipitates from precisely measured reagents. Regular decanting by
pouring off drags a small amount of precipitate with the last few drops of solvent. Accidental tipping is easy, and losses are not consistent from
batch to batch.
I tried coffee filters with the idea that I could dry them and weigh them with the preciptiptiate (tare weight). I hoped to get a very accurate
measurement of precipitate weight; but they take a very long time to gravity filter as the pores plug and quite a bit of precipitate will climb up the
paper sides like chromotography paper and sometimes get out of the filter.
So, I bought this (typical) glass fritted buchner funnel from ebay seller laboratoryglassware:

I am quite happy with the speed and amount of water that is removed by a Buchner funnel with glass frit and drip tube (to prevent fluid going to
vacuum pump or asperator). It is SO much faster than a coffee filter. I also like being able to see the chemicals being processed, which would be
hard with a porcelain funnel; Not to mention, that glass filters come with a standard taper joint; whereas porcelain ones I have seen don't. Rubber
bungs are easily attacked by ketones and other solvents, so a glass joint is safest.
The only down side is that the glass frit also captures some of the precipitate, and weighing the entire buchner funnel with precipitate isn't
practical. Even using filter paper discs is very difficult because removing them without tearing is nearly impossible once moistened on top of glass
frit. Getting the precipitate to total dryness on top of glass frit causes problems. I haven't seen a glass vacuum filter with perforations instead
of glass which would make removing the filter easier. I am curious as a side question if anyone makes glass joint Buchner funnels with perforations
instead of glass frit.
But:
The ideal solution would be a cheap pre-made "Buchner" wand (inverted buchner funnel) that can be inserted into an Erlenmeyer flask or a vial/test
tube and allow the waste solution to be pulled out with a vacuum line while leaving solid precipitate in the flask. Since the hole size is small,
only a microscopic amount of precipitate would be lost in the frit and it would be repeatable. eg: I'm wanting a decanting aid to prevent the
accidental loss of precipitate which happens at the end of a decantation when the last drops are trying to be removed from the solution.
The following drawing is my idea to minimize loss of precipitate for testing small batches of chemistry. ( 8 mL of solution or so can easily be
removed through a small glass tube with inner diameter of 0.5 to 3 millimeters...and glass frit would make a good filter just like in Buchner funnels.
)
I have a pretty good idea how to make one, using capillary tubing and/or normal borosilicate glass tubing; but I'm curious if something low cost
already exists. If not, then I will share my ideas about how to make the glass frit; and perhaps someone can help me with ideas to improve it (public
domain ideas) making it chemically inert as possible while not totally plugging the tube.
[Edited on 29-12-2017 by semiconductive]
|
|
|
happyfooddance
National Hazard
Posts: 530
Registered: 9-11-2017
Location: Los Angeles, Ca.
Member Is Offline
Mood: No Mood
|
|
I think you might be confused as to what a "buchner" funnel is. What is in the picture above is a "fritted disc" filter, or just "frit". They come in
different porosities and are generally used for filtering solutions.
"Buchner" funnels are what you described that is similar, but with perforations, not fritted glass. They are used with a filter just small enough to
fit in the base, and are used to separated crystallized product from a supernatant liquid.
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Still, this seems like an interesting idea. How would you prevent clogging of the tube?
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
Something similar was used in a low temperature recrystalization of iodopropyne.
http://orgsyn.org/demo.aspx?prep=v93p0245
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
I might have some small tubes with frits on the end if you are interested. They are not as small as you show, but not bad. There was a company
making needles with round ends and micro holes in them for the same purpose. but not sure if they are still available, I think they clogged too much
and were abandoned.
Another simple solution is to use membrane filters, made of teflon or similar materials, which go into special frames that hold them in place, with a
glass frit below the membrane, which allows solvent through, but the filters are good to 0.5 micron of so. I have scads of the larger holders, in 47
mm size, maybe a few of the smaller ones, which are about 20-22 mm diameter. That would likely be a good solution for you. I don't have the
membranes, however, but they are not too expensive.
[Edited on 30-12-2017 by Dr.Bob]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
| Quote: | Quote: Originally posted by happyfooddance  | | I think you might be confused as to what a "buchner" funnel is. What is in the picture above is a "fritted disc" filter, or just "frit". They come in
different porosities and are generally used for filtering solutions. |
|
That's possible; but the glass frit filters are sold on ebay under the name "Buchner" funnel from pretty much all sellers. Most sellers do not put
"fritted disc" as part of the title. In general, the name Buchner is applied to any filter which uses vaccum.
So, I thought the idea of vacuum filtration in general was attributed to Buchner.
Now that I'm searching again ... I've found several glass Buchner funnels with glass "pore" plates and ground glass joints; one of them is ironically
made by the manufacturer I bought my fritted disc funnel from six months ago, when there were no glass buchners for sale on ebay for some odd reason.
I even asked at the time. *sigh*
[Edited on 30-12-2017 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Glass frit also clogs, it's just doesn't completely clog in a useable funnel.
Vacuum is a big help in getting fluids past clogs.
There are also two easy ways to reduce / prevent clogging. The first is just to move the frit away from the tip, either partway through the tube or
at the other end. This gives gravity time to work on the precipitate before it gets to the fritted filter. If the vacuum is reduced, so that the
liquid is pulled up reasonably slow ... that will have a strong effect. The second way to reduce clogging, is to make the tube have a bubble expanded
glass area, that forces the solvent to slow down compared to the tube and travel along an arc before exiting through a fritted area. That acts a bit
like a centerfuge, and if the frit is in the middle; then is aids in separating the two. Some fluid may remain in the bubble at the end of
suctioning, but it could be sloshed back into the vial after turning off the vacuum.
As long as precipitate isn't lost, it doesn't hurt to push fresh solvent with no contaminants back through the wand in reverse to dislodge any
precipitate in the tube.
I've manually decanted vials of liquid using an eye dropper. Basically, I pull up a dropper full of fluid, wait a moment for the precipitate to
settle and then release one drop of fluid back-wash with all the precipitate. I'm able to get far more liquid out without loosing precipitate this
way (compared to dumping sideways), but it's a tedious process.
A third idea, is to put a teflon buoy on the tip so that the wand floats at the surface and only lowers as the solvent is removed. Since the
precipitate goes to the bottom of the vial, this would automatically keep it from clogging until there is only moisture inside the precipitate.
Again, if I loose a small and consistent amount of precipitate; that at least can be estimated as a systematic error and corrected for. It's when I
loose random amounts of precipitate that I become frustrated.
Unclogging frit is usually as easy as running a strong acid or solvent through it to dissolve the precipitate. If the frit manufacture is cheap
enough, then the tubes can become disposable when irreparably damaged. Glassware tubing is usually less than a dollar per piece.
|
|
|
happyfooddance
National Hazard
Posts: 530
Registered: 9-11-2017
Location: Los Angeles, Ca.
Member Is Offline
Mood: No Mood
|
|
I think I understand what you are getting at. You are looking for a way to decant without disturbing settled precipitate. A frit will work, a
capillary tube works better. I have a small army of different tubes, pp, hdpe, pvc, all with fittings and clamps to hook up to glass adapters.
Sometimes I use 1/4" hpde just by itself, usually to vacuum-fill flasks. You can even use a stopcock (I use a brass one for gas lines) to adjust or
stop flow. This only works if there is no air between the stopcock and where you are drawing fluid. I have some 24/40 thermometer adapters with a
vacuum barb, with these you can use single neck flasks as your "wash" bottle. Capillary tubes work best because you can very precisely control the
point where you draw up fluid.
What you did with the eyedropper is not that uncommon, usually an appropriately sized pipette is used, so as to make it less tedious. You can shove
some glass wool or rayon in there too, to help filter.
|
|
|
happyfooddance
National Hazard
Posts: 530
Registered: 9-11-2017
Location: Los Angeles, Ca.
Member Is Offline
Mood: No Mood
|
|
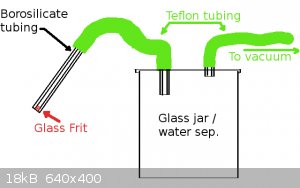
Sometimes I don't even intersperse a wash bottle, when I am removing mostly aqueous things that are just going down the drain anyways. This little
set-up gets a lot of use just for it's convenience.

|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
| Quote: Originally posted by happyfooddance | Sometimes I don't even intersperse a wash bottle, when I am removing mostly aqueous things that are just going down the drain anyways. This little
set-up gets a lot of use just for it's convenience.
|
I can well imagine! That's a good idea.
Yes, you have the right idea.
I bought a bunch of borosilicate capilary tubing with a 0.3mm bore. I also got a handful of 1.9 ID, 3.8mm OD borosilicate glass tubing meant for
melting and bending. I was intending to make a constantin/copper thermocouple thermometer (sealed) with it since stainless steel sheathed
thermometers and aquia regia don't get along.
I didn't think of trying to modify a pipette, like you suggested earlier.
I have never seen a thermometer adapter with a vacuum take off. Is there a special name for them ? The lowest cost thermometer like adapter I have
seen is a $30 ACE GLASS "5261-56" adapter but it doesn't have a drip tube, tightening nut or sheath for the thermometer ... so some of the fluid would
go from the inlet into the vacuum pump.
When I made the drawing, I was thinking to use a glass canning jar with vacuum seal lid.
Marine epoxy is good enough to hold the glass inlet tube in the lid, and a little silicone to seal the lid against extremely corrosive liquids is fine
in a wash bottle. I have one of those freezer bag vacuum sealer pumps from Reynolds corporation; So I was going to just ppunch a hole for the vacuum
pump suction head and put a small bit of sodium bicarbonate into the pumps nozzle to neutralize any corrosive acid gasses that manage to evaporate out
of the wash bottle.
The other way I was thinking to do it, is to take one of my $1.00 hollow glass flask plugs and drill two holes in it, insert the glass tubing and then
fuse the two together with a mapp gas torch. Then I could use any kind of vacuum pump ( faucet based ones) with that plug on an Earlenmeyer flask.
(24/40.)
I have seen cheap adapters for about $8 from china like this one. Search for nanshin glass on ebay.

It looks like it's meant for typical 1/4 inch hose, I think. The glass tubing I have isn't the same size, so I still would have to melt some kind of
1/4 vacuum barb onto the glass tubing I have to make a wand with.
The only thing I don't like about this plug is that the bottom of the drip tube is flat. I really prefer the drip tube to be cut at an angle with a
sharp point. That makes the liquid drip off cleaner and faster rather than hanging on to the adapter when it gets pulled out of a Earlenmeyer flask.
The dead space inside the plug on the vacuum adapter is really good, because it's trivial to make silicone membranes using hardware store caulk for
fish tanks. Silicone caulk will pass gasses but not liquids. That dead space is also a good spot to put neutralizing bicarbonate crystals as an
extra safety measure for mechanical pumps.
There are also "glass pourous adsorbing tubes" which are pretty cheap, $6 including shipping from wondernidaye on ebay. Those would require me to
remove the tube every time I wanted to empty them out, though. So, the flexible tubing would receive a lot of wear and tear.
The only thing I could wish for, more than the earlenmeyer wash bottle, would be a teflon valve to adjust or block the vacuum to the wand. However,
there's only two styles of vacuum transfer adapters with valves that I see ... those with a single valve "three way", and no drip stem at all; and
those with two separate valves for both inlet and outlet that are substantially more expensive ($25+ each.)
Teflon is even more expensive than glass valves, but I'm not sure how troublesome glass is with getting stuck as a valve.
Those prices seem pretty high for something that is so simple and can probably be made in a few minutes using cheap borosilicate glass plugs and a
propane torch. I mean, a properly shaped tube could just have a magnetic stirrer dropped into it as the valve plug; and then use a rare earth magnet
to open and close it in a totally sealed pipe.
[Edited on 4-1-2018 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
| Quote: Originally posted by Dr.Bob | | I might have some small tubes with frits on the end if you are interested. They are not as small as you show, but not bad. |
I'm curious, what is the diameter of the tubing ?
My thought was just to use 3.6mm lab glass tubes, or small capilary tubing ... and then take water-glass with borax to plug the tip.
There are several ways to adjust the density of the glass and make it porous while fusing the sodium silicate into borosilicate glass inside the tube
at the same time.
| Quote: |
Another simple solution is to use membrane filters, made of teflon or similar materials, which go into special frames that hold them in place, with a
glass frit below the membrane, which allows solvent through, but the filters are good to 0.5 micron of so. I have scads of the larger holders, in 47
mm size, maybe a few of the smaller ones, which are about 20-22 mm diameter. |
I have 50mL pyrex Earlenmeyers, and I think those are 19/22 ground glass necks. It's the American joint, so that should be 19-1/10*22/2 = ~18mm at
the narrowest.
All the other Earlenmeyers I have are 24/40 Chineese; so those would be 24 at the widest, and 24 - 40/10 = ~20mm at the narrowest. Do you have a
photo of the smallest holders?
Who sells the Teflon membranes? I know how to make silicone membranes, but silicon has about a 1 to 2 micron pore size naturally. Teflon sounds like
it would be better for inertness if the price is decently low.
I'm also trying to filter inside vials and avoid transferring the precipitate to avoid spills and transfer wetting losses; my rack of vials is shown
in this other thread and I don't think any of those filters will work, correct?
https://www.sciencemadness.org/whisper/viewthread.php?tid=79...
The reason I'm using these tubes is that I can mark them in bulk with tare weights, mix the experiment in several variations, and then let them sit in
a rack for two to three days; eg: off my workbench and fumbling hands. The vials don't hold much chemical, so even tiny losses will skew the results
of weighing precipitate. If the loss is a consistent systematic error, then it can be compensated for. But having 24 to 48 vials let me do longer
term experiments with precise results in a limited space.
[Edited on 4-1-2018 by semiconductive]
|
|
|
happyfooddance
National Hazard
Posts: 530
Registered: 9-11-2017
Location: Los Angeles, Ca.
Member Is Offline
Mood: No Mood
|
|
https://www.amazon.com/gp/aw/d/B01ASDDN34/ref=mp_s_a_1_10?ie...
This is a link to the adapter I was talking about.
I know why you want to implement a stopcock, but it might not be as convenient as you imagine, when you get one and set it up to your line and try to
use it you will see the difficulty it presents, with little benefit to the apparatus.
... Especially for small amounts, the reason being, as I mentioned briefly before, that the stopcock will not stop or even considerably slow flow,
until the capillary and tubing are filled with liquid completely to the point of the stopcock. It will continue to suck up liquid after closed,
because of the partial vacuum that exists in your tubing. This is especially frustrating if you need to only suck up a small amount, or repeatedly
start/stop at the end of a decantation. I am just warning you before you spend good money on an expensive stopcock for this purpose, although that is
a great thing to have for other purposes.
All in all, you will have better control by manually removing a capillary tube as opposed to fidgeting with a stopcock, guaranteed. You can slow the
flow by adjusting the water flow to your aspirator, if that is your vacuum source. The aspirator I showed earlier is Nalgene brand, it has a valve
which prevents backflow if pressure is reduced. $16 at Carolina Biological.
[Edited on 1-4-2018 by happyfooddance]
[Edited on 1-4-2018 by happyfooddance]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Thanks for the link. That's very helpful to get an idea of how to do things. That plug design would be simpler, because the Teflon tubing can easily
be sliced at an angle (rather than flat ended) and then just inserted past the vacuum port. Once it's below the port, it's as good as the plug I
showed ... but without having to use larger teflon tubing to fit over the glass gas fitting. eg: > 1/4 inch tubing.
Unfortunately the European & Chineese glassware companies aren't selling an equivalent part. All my larger flasks have the European 24/40 joint ..
which measures the diameter at the top of the plug. However, American companies, following Pyrex (TM) and marked TS, measure the plug diameter at the
middle of the plug or below. So, I can put a European plug into an American flask, and it will go all the way in and seat partway into the neck.
But, if I try to put an American plug into a European socket ... it won't even go halfway in and often doesn't seal well.
I looked at gas wash bottles that were shown in the sub categories of the same link you gave, and for a stupid test-tube sized one with the plug and
vacuum port costs $30. So, I think I'll have to look a little more.
Thanks for the warning. You're right. A manual valve won't be useful.
I hadn't thought about that carefully. However, I still eventually want to regulate the vacuum because I can forsee some of the experiments that I
want to do ... and the problems with techniques that require me to be quick with my hands, or extra careful.
1) I only have two hands and am sometimes clumsy.
2) Many of the chemical experiments I want to do will likely be ruined (or possibly explode) if exposed to air or heat, so I need to have a source
port for an inert gas.
3) I will probably need to supply either butane or CO2 gas where the chemical is, while trying to vacuum out the liquid on top of it requiring a lot
of dexterity. Doing the experiment inside a ziplock bag full of butane would waste a lot of butane, make handling very difficult and spills likely.
4) I can't turn the vaccum pump itself down, so I'm wanting some way to keep the suction low. eg: a needle valve or other method to regulate the
vacuum level. After thinking about it, probably an electronic method would be better than a needle valve since it would also free up my hands if I
made it automatic.
I think the vacuum lag due to air in a tube could be reduced by simply using a very tiny inner diameter tubing. Then there is very little air inside
the tube in the first place. I'm not sure it will work, and I really appreciate your warning... so, I won't waste a lot of time on larger tubes.
The capillary glassware I own is 0.4mm ID, and 0.9mm OD. , (eg: 300 micron ID). It's cheap and disposable if it plugs.

I looked at teflon tubing, and there is disposible REP-RAP printer PTFE tubing for about $1.00/for every 300mm length (about a foot.) It also comes
as small as 0.3mm ID. ( Slightly smaller than a capillary tube I.D. !!!!) So, I'll expeiment with that and see if it still can pull sufficient
vacuum to pull fluid up the tube. I've been told that as tube diameters get small, that the vacuum actually transmitted by the tubes is reduced
significantly. This could be a bad or good thing, because if it acts as a throttle to slow down the rate of suction from a vial ... it would
naturally make the system less likely to suck up precipitate.
[Edited on 8-1-2018 by semiconductive]
|
|
|
|








