DrDevice
Hazard to Self

Posts: 75
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
Tosylation of ethanolamine (??)
I have been experimenting with tosylation of ethanolamine, expecting that both the alcohol and the amine components of the ethanolamine will react to
give a "ditosylate".
I dissolved 38.65g (0.2mol) of tosyl chloride (TsCl) in 80ml tetrahydrofuran (THF), and added 30g (0.3mol) of triethylamine. This was then cooled
using an ice bath to below 5C.
6.3g (slightly more than 0.1mol) of ethanolamine was added drop wise with magnetic stirring, ensuring the temperature did not rise above about 7C.
Once all the ethanolamine was added, the solution was stirred at room temperature overnight.
The insoluble white coloured triethylamine hydrochloride was filtered. Some pressing of the solids was required to extract as much liquid as possible.
The solids and filter paper were washed again with more THF to remove as much product as possible.
The resulting liquid was heated to 80C under reduced pressure (0.1atm), to remove as much solvent as possible. On cooling, I was left with 38.1g of a
viscous, pale yellow liquid. If it is what I think it is, structure is:
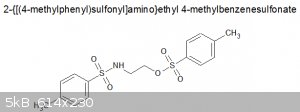
MW = 369.46, so 0.1mol. I'm sure there must be contaminants in there, but yield is pretty good.
Assuming of course that I'm not fooling myself... Comments or criticisms pls? Also, any suggestions on (accessible) quantitive or qualitative tests I
can do to verify this result? Can I
Next, I want to substitute the "alcohol" end with iodine, leaving the "amine" end untouched. Suggestions? Will sodium iodide dissolved in acetone
suffice?
|
|
|
morsagh
Hazard to Others
Posts: 187
Registered: 20-2-2014
Member Is Offline
Mood: No Mood
|
|
Most easy test is to try TLC and compare to ethanolamine and TsOH. Try colouring with iodine, every of these should be visible.
|
|
|
DrDevice
Hazard to Self
Posts: 75
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
Can you (or anyone...) suggest an introductory text/website on TLC (or eg paper chromotography) with examples that I can use for checking my reaction?
Best to assume I know nothing about the subject 
|
|
|
morsagh
Hazard to Others
Posts: 187
Registered: 20-2-2014
Member Is Offline
Mood: No Mood
|
|
You just take your TLC plate, on the bottom you make a line with pencil, there you will make one point with your product, next to it another points
which are your reactants or possibly byproducts. Then just compare. https://en.wikipedia.org/wiki/Thin-layer_chromatography on wiki it is written very good. For your compounds i would use 1:1 mixture of petrolether
to ethyl acetate. Then just adjust the ratio. One more tip, if you are getting a long line, then your solvent is just too polar.
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Just a brain fart; Can't you hydrolyse with a know quantity of NaOH and then titrate with acid to see how much base was consumed? Two mol NaOH per mol
of compound would mean a ditosylation
|
|
|
morsagh
Hazard to Others
Posts: 187
Registered: 20-2-2014
Member Is Offline
Mood: No Mood
|
|
I think if there is any unreacted tosyl chloride present, it won´t be distilled so it will remain in his product so titration will always show
"positive result". I am not sure...
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
Likely worked at least most of the way. I think the NaI in acetone should work, if not DMF instead, but acetone is better for solubility of the
salt. You will likely need to reflux it a while.
|
|
|
DDTea
National Hazard
Posts: 940
Registered: 25-2-2003
Location: Freedomland
Member Is Offline
Mood: Degenerate
|
|
In my experience with 1,8-octaneditosylate, yellow oil meant "reaction did not go to completion." Beautiful white needles were the measure of
success. Run the TLC and see how many products you have. You can always re-subject the yellow oil to the original reaction conditions (i.e., TsCl ,
NEt3, in THF) to improve your yield. Otherwise, starting with an excess of the TsCl here may be useful.
Depending on where you got your TsCl and its age, it may need to be cleaned up by a simple recrystallization. Two methods for doing so are as follows
(I use the first, never tried the second):
| Quote: | Tosyl chloride (CAS NO. 98-59-9) is purified by dissolving (10g) in the minimum volume of CHCl3 (ca 25mL) filtered, and diluted with five volumes
(i.e. 125mL) of pet ether (b 30-60°C) to precipitate impurities. The soln is filtered, clarified with charcoal and concentrated to 40 mL by
evaporation. Further evaporation to a very small volume gave 7g of white crystals which were analytically pure, m 67.5-68.5 °C. (The insoluble
material was largely tosic acid and had m 101-104°C).
It is also crystd from toluene/pet ether in the cold, from pet ether (b 40-60 °C) or benzene. Its soln in diethyl ether has been washed with aqueous
10 % NaOH until colourless, then dried (Na2SO4) and crystd by cooling in powdered Dry-ice. Tosyl chloride has also been purified by dissolving in
benzene, washing with aqueous 5% NaOH, then dried with K2CO3 or MgSO4, and distd under reduced pressure and can be sublimed at high vacuum.
|
From http://www.lookchem.com/Chempedia/Chemical-Technology/Labora...
"In the end the proud scientist or philosopher who cannot be bothered to make his thought accessible has no choice but to retire to the heights in
which dwell the Great Misunderstood and the Great Ignored, there to rail in Olympic superiority at the folly of mankind." - Reginald Kapp.
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
DrDevice:
Your reaction work up was incomplete. After filtration of the triethylamine hydrochloride and removal of THF, your CRUDE product needs to be taken up
in a suitable solvent and subjected to an acid-base wash regimin. This is the minimum purification that you should do. As others have pointed out, you
also need an analytical method to assess purity. The hydroxide hydrolysis/titration suggestion is going to tell you much if anything. Finally, your
target compound is in all likelihood known. What do you know about it?
AvB
|
|
|
DrDevice
Hazard to Self
Posts: 75
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
Thanks all for the feedback so far. I don't have TLC plates on hand, but attempted to use paper chromatography, with iodine as an indicator ie putting
the paper into a beaker with a small amount of solid iodine to sublimate - thanks morsagh for chromatography suggestions.
The results were very dodgy, but there was a line in the "product" that was at the same displacement as the ethanolamine line, so I'm assuming an
excess of ethanolamine.
I've re-dissolved the original product in THF and added more triethylamine and extra TsCl such that (compared to original amounts) would be 2.05
equivalents.
This is stirring now. I'm not seeing any white hydrochloride ppt however.
AvB, any suggestions on an acid-base wash workup?
And as for the final product, what do I know about it? I have had a look through the readily accessible literature for "tosylation of ethanolamine",
but no obvious results.
|
|
|
morsagh
Hazard to Others
Posts: 187
Registered: 20-2-2014
Member Is Offline
Mood: No Mood
|
|
I don´t know how it is with solubility of Ts esters in water but amides should be insoluble. TsCl should hydrolyse and TsOH dissolve (violent
reaction be careful, lot of HCl evolved, work in fumehood). Ethanolamine is soluble too.
|
|
|
DDTea
National Hazard
Posts: 940
Registered: 25-2-2003
Location: Freedomland
Member Is Offline
Mood: Degenerate
|
|
I recommend more like 3 - 4 equiv TsCl to 1 equiv ethanolamine. After the initial exotherm, try heating/refluxing for a bit. There really are no
expected side reactions here, so you can force it to completion.
"In the end the proud scientist or philosopher who cannot be bothered to make his thought accessible has no choice but to retire to the heights in
which dwell the Great Misunderstood and the Great Ignored, there to rail in Olympic superiority at the folly of mankind." - Reginald Kapp.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
DrDevice, I don't think you chose suitable conditions. Yes, triethylamine and TsCl can be used to tosylate alcohols, but preferably in
dichloromethane. For some reason dichloromethane works best with either pyridine or triethylamine for the reaction of sulfonyl chlorides with
alcohols, or poorly nucleophilic amines and anilines. Other solvents are either substantially inferior or do not work (with the exception of
pyridine). Also, triethylamine is avoided in favor of pyridine when sulfonating primary alcohols because alkyltriethylammonium salts tend to form with
excess triethylamine unless the reaction is properly monitored.
Since N,O-ditosylated ethanolamine is a crystalline solid, you should do two obvious things. First you should check the purity with TLC and then if
still optimistic crystallize it from some solvent (e.g., methanol or ethanol/water). But without TLC plates... (I don't understand why you even
started with organic reactions without first buying some TLC plates).
Why do you want to substitute the tosyloxy for the iodine? There are only very few reactions where this would be required. If you want to make
N-tosylaziridine, then you just treat the product with NaOH. Also, if you want to alkylate something, you can also just use the O-tosylate. There is
no need for iodine exchange.
Quote: Originally posted by DrDevice  | | And as for the final product, what do I know about it? I have had a look through the readily accessible literature for "tosylation of ethanolamine",
but no obvious results. |
I don't know how hard have you looked, but there are several examples of this reaction in the literature. Since you say you don't have TLC plates, the
least you should do is follow a verified procedure.
| Quote: Originally posted by morsagh | | TsCl should hydrolyse and TsOH dissolve (violent reaction be careful, lot of HCl evolved, work in fumehood). Ethanolamine is soluble too.
|
The hydrolysis of TsCl is certainly not a violent reaction. It proceeds quite slowly actually and TsCl is not that easy to remove with just an
extraction based work up.
[Edited on 4/1/2017 by Nicodem]
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
I like using just 2.1 or so equiv of TsCl, too much will be a pain to remove. I do agree that DCM would be a better solvent, but it might go in THF.
A small amount of pyridine, or better yet, DMAP, will help catalyze the reaction, DMAP does not smell so bad, and would partition into the acid wash
if you take the crude reaction (in DCM) and wash with 1N HCl and then sat'd bicarb to remove excess SM and salts. While TEA might not be ideal, it
should work OK, it is also easy to find, and will come out in the water wash. Certainly recrystallizing the product will help if it is not pure, but
always better to drive the reaction to completion if at all possible. Pushing the crude material through a plug of silica gel in hexanes to ethyl
acetate would also likely clean it up enough to crystallize.
I found the same article that Nicodem found, I think, it has an NMR of the product in the supplimentary material, might have other info in the paper
itself, but I can't get to that right now. But their procedure is likely a good starting point. You should be able to get the article via a library
or other sources.
http://www.tandfonline.com/doi/full/10.1080/10426507.2015.10...
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
I agree with Nicodem that the solvent of choice for triethylamine tosylations is dichloromethane. Why this is so I cannot say.
DMAP often leads to side products with acid chlorides which involve addition of the acid chloride to the pyridine ring. This is documented but the
reference is not currently at hand. DMAP catalysis with acid chorides is almost never any better than use of plain pyridine.
Here is the experimental data from the paper cited by Dr Bob:
N-[2-[(p-Tolylsulfonyl)oxy]ethyl]-p-tolylsulfonamide
Monoethanolamine (12.00 g, 0.197 mol) was dissolved in anhydrous pyridine (15 mL) and
cooled to 0 oC in a bath of ice and salt. Tosyl chloride (80.00 g, 0.419 mol), finely powdered,
was suspended in pyridine (50 mL) and cooled to -50 oC. The ethanolamine solution was added
while the temperature was maintained below 10 oC for 30 min. The mixture was kept at room
temperature for 6 h and then at 4 oC overnight. The mixture was poured, with stirring, on crushed
ice (500 g) and acidified with glacial acetic acid (20 mL), whereupon the oily material solidified.
The solid was crushed to a slurry in water, collected, and washed with water. The product was
dried in desiccator over P2O5 in vacuum. Recrystallization of the crude product (69 g) from hot
chloroform (50 mL) and addition of cold CCl4 (200 mL) gave 1 as a white powder (55.00 g, 76
%), mp 87-88oC, mp 86-87 oC.20 1H NMR (400.130 MHz, CDCl3): δ 2.41 (s, 3Н, Me), 2.44 (s,
3Н, Me), 3.18-3.22 (m, 2Н, CH2), 4.03 (t, J= 5.3 Hz, 2Н, CH2), 5.00 (br s, 1H, NH), 7.27 (d, J=
8.3 Hz, 2H, p-C6H4), 7.33 (d, J= 8.3 Hz, 2H, p-C6H4), 7.68 (d, J= 8.3 Hz, 2H, p-C6H4), 7.73 (d,
J= 8.3 Hz, 2H, p-C6H4) ppm; 13C NMR (100.613 MHz, CDCl3): δ 21.4, 21.6 (2Me), 42.0, 68.7(2CH2), 127.0, 127.9, 129.8, 130.0, 132.2, 136.6, 143.7,
145.2 (2C6H4) ppm
AvB
|
|
|
brubei
Hazard to Others
Posts: 188
Registered: 8-3-2015
Location: France
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Nicodem | | But without TLC plates... (I don't understand why you even started with organic reactions without first buying some TLC plates).
|
so true ...
[Edited on 5-1-2017 by brubei]
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
| Quote: Originally posted by AvBaeyer | | DMAP often leads to side products with acid chlorides which involve addition of the acid chloride to the pyridine ring. This is documented but the
reference is not currently at hand. DMAP catalysis with acid chorides is almost never any better than use of plain pyridine. AvB
|
I will agree that there are some cases where DMAP is not as good as pyridine, maybe even more so for carboxlic acids, but I have made over 2000
discrete sulfonamides with DMAP, most using DCM (DCE also does quite well, better for some cases) as the solvent and TEA as the base, and most were
>95% pure straight from the reaction, after a simple workup. If you use DMAP on resin (either catalytically or as the main base at 1.5 eq), that
is even better, as it just filters away, and you can use a smaller amounts of TEA (catalytic, maybe as little as 0.1 to 0.5 eq), it acts as a proton
shuttle in that case, and the DMAP on resin both activates the sulfonyl chloride and acts as a base. One of the best reactions I have ever used in
parallel synthesis. Best way is react sulfonyl chloride with very well dried DMAP resin in dry DCM for an hour or so at RT, then add the amine or
alcohol along with some TEA. Let them stir overnight and filter.
[Edited on 5-1-2017 by Dr.Bob]
|
|
|
DrDevice
Hazard to Self
Posts: 75
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
Over the last few weeks I have been looking at this reaction again. I have purchased some TLC plates, and currently getting familiar with how to use
this process. Encouraging results, but that's not the main reason for this post.
I have pursued two separate approaches to the tosylation, one based (very loosely) on the procedure provided by AvBaeyer, the other in DCM,, as others
have commented. Basically:
Method 1 involved dissolving 30.54g (0.16mol) TsCl in 60ml DCM with 23.2g (0.23mol) triethylamine. This was chilled in an ice/salt mix, and 4.64g
(0.076 mol) ethanolamine added drop wise ensuring temperature did not rise above 10C. This was left stirring overnight at room temperature. A yellow
slurry formed. This was poured onto 200g of ice with 20ml acetic acid. Once the ice melted, the organic and aqueous layers were separated, and the
solvent evaporated. The remaining solid was re-dissolved in boiling ethanol (it is not very soluble in EtOH), and re-crystalized to a white powder.
Yield 7.5g. I did muck around a bit trying different solvents, so I probably lost a bit in the process.
Method 2 was closer to the one in the procedure provided by AvBaeyer. 21.91g (0.115mol) of TsCl, finely ground, was added in small portions to 60ml of
rapidly stirring morpholine at -4C. A suspension is formed, and some dense white vapour is evident when the TsCl is added. 3.3g (0.054mol) of
ethanolamine is dissolved in 20ml of morpholine, and added dropwise to the TsCl suspension, keeping the temperature below 10C. This was stirred for
0.5hr at 0C, then 24 hours at room temperature.
The slurry/solution was poured onto 200g of ice with 20ml acetic acid. The white precipitate was gathered by filtration and dried in air. The ppt was
dissolved in warm chloroform. Some remaining water was evident so I dried with sodium sulfate, then filtered whilst hot. On cooling, the white
precipitate re-formed in the CHCl3. I have taken some of this precipitate and dissolved in boiling IPA. The product recrystalized in beautiful long,
fine needle-like crystals. Total yield approx. 16g (I am still drying the IPA crystalized product.
I used morpholine rather than pyridine as it is easier for me to obtain - and I had some. I reasoned that it was a relatively weak base like pyridine,
so it might work.
So all this sounds good in that I am getting *something* consistent with either approach. HOWEVER, my product in each case has a melting point of 145C
- 147C, vastly different from the 87C-88C quoted in the experimental results from the paper.
So any suggestions? I'm ending up with what looks like the same product from each approach, but doesn't agree with the quoted MP. Is it likely that I
have ended up with something so completely different by a different solvent choice?
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DrDevice |
I used morpholine rather than pyridine as it is easier for me to obtain - and I had some. I reasoned that it was a relatively weak base like pyridine,
so it might work.
|
Morpholine is some three orders of magnitude more basic than pyridine, and significantly more nucleophilic also. The other factor is that it is a
secondary amine, and so reacts with the TsCl to form a stable sulfonamide, rather than the reactive N-p-toluenesulfonyl)pyridinium
intermediate you get with pyridine which facilitates the desired reaction with the amine substrate.
All in all I'd say it was a poor substitution to make.
Edit: 4-tosylmorpholine is reported to have a mp of 142 to 144 *C, and the N-(2-hydroxyethyl)-p-toluenesulfonamide is reported to
have mp 55 to 57 *C. Not sure where you got 87 to 88 *C from?
[Edited on 2-2-2017 by DJF90]
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
My post of the experimental procedure explicitly stated the use of pyridine. I do not believe it is unreasonable for anyone running a tosylation
experiment to know the difference between pyridine and morpholine with regard to chemical properties. That's pretty basic stuff. It's possible that
N-methylmorpholine, a common acylation catalyst, could replace pyridine.
DFJ90: The 87-88*C mp is for the bis-tosylated compound. See my post above.
AvB
|
|
|









