| Pages:
1
..
10
11
12
13
14 |
subsecret
Hazard to Others

Posts: 424
Registered: 8-6-2013
Location: NW SC, USA
Member Is Offline
Mood: Human Sadness - Julian Casablancas & the Voidz
|
|
I noticed that someone posted a reference where benzene was produced from toluene, HCl, and AlCl3, which causes some of the methyl groups
to migrate between benzene rings, forming xylene, mesitylene, etc.
Wouldn't a longer reflux time work to produce benzene? After all, if this is the reverse of a Friedel-Crafts, chloromethane should be produced, which
would boil off at a low temperature. This should drive the equilibrium to the side with the benzene.
I didn't see the original PDF, so here it again for consideration.
Attachment: The Friedel-Crafts reaction Part III.pdf (287kB)
This file has been downloaded 1379 times
Fear is what you get when caution wasn't enough.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
After a few attempts at synthesising Benzene from PVC pyrolysis, i'm giving up.
Not one experiment yielded any useable benzene.
You mostly get HCl, Carbon, and a small amount of brown gunk that makes a huge mess of the glassware.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by aga  |
You mostly get HCl, Carbon, and a small amount of brown gunk that makes a huge mess of the glassware. |
what about the large number of carcinogenic chemicals like dioxins etc. 
but no one tried the method posted by this guy,maybe it was too crazy
http://www.sciencemadness.org/talk/viewthread.php?tid=508
[Edited on 2-1-2015 by CuReUS]
|
|
|
subsecret
Hazard to Others
Posts: 424
Registered: 8-6-2013
Location: NW SC, USA
Member Is Offline
Mood: Human Sadness - Julian Casablancas & the Voidz
|
|
As stated by aga, the PVC method isn't practical. But while on the topic of dioxins, thermolysis may be a good way to get rid of these compounds. Run
the gas stream into the bottom of a hot fire to decompose any undesirable waste.
Fear is what you get when caution wasn't enough.
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Awesomeness |
As stated by aga, the PVC method isn't practical. But while on the topic of dioxins, thermolysis may be a good way to get rid of these compounds. Run
the gas stream into the bottom of a hot fire to decompose any undesirable waste. |
Except that "backyard" burning of organochlorine containing waste is the single largest source of environmental dioxins. They aren't necessarily
formed as the material burns and more fire won't fix it. With regards to incinerators (formerly the major source), wiki has this to say:
"In incineration, dioxins can also reform or form de novo in the atmosphere above the stack as the exhaust gases cool through a temperature window of
600 to 200 °C. The most common method of reducing the quantity of dioxins reforming or forming de novo is through rapid (30 millisecond) quenching of
the exhaust gases through that 400 °C window. Incinerator emissions of dioxins have been reduced by over 90%"
|
|
|
maleic
Harmless
Posts: 20
Registered: 24-12-2014
Member Is Offline
Mood: No Mood
|
|
Propylene and benzene under the catalysis of aluminium trichloride, from 80 to 90 degrees Celsius for hydrocarbon reaction, get the isopropyl benzene.
Isopropyl benzene with air in the 100-120 degrees Celsius, and 100-400 kpa pressure isopropyl benzene oxidation generating hydrogen peroxide.
Isopropyl benzene with sulfuric acid hydrogen peroxide under 60 ℃ atmospheric pyrolysis for phenol and acetone.
|
|
|
subsecret
Hazard to Others
Posts: 424
Registered: 8-6-2013
Location: NW SC, USA
Member Is Offline
Mood: Human Sadness - Julian Casablancas & the Voidz
|
|
Just a note:
JB Weld will does not withstand the effects of dry distillation of sodium benzoate and sodium hydroxide. I sealed a copper tube into a paint can with
it, but several minutes after I applied heat, it began to flake and leak benzene vapor.
Fear is what you get when caution wasn't enough.
|
|
|
DFliyerz
Hazard to Others
Posts: 241
Registered: 22-12-2014
Member Is Offline
Mood: No Mood
|
|
I made benzene with sodium benzoate and sodium hydroxide too, and I think I have an idea of what causes the orange/yellow colors. After researching
for a little while, it seems that the yellow/gold could be caused by chrysene, and the orange caused by tetracene.
|
|
|
maleic
Harmless
Posts: 20
Registered: 24-12-2014
Member Is Offline
Mood: No Mood
|
|
Sodium benzoate in soda lime heating conditions can generate sodium carbonate and calcium carbonate and benzene, due to the heating condition, so the
temperature is not very high, so the sodium carbonate and calcium carbonate does not decompose, and the boiling point of benzene is 353.25 K (80.1 DEG
C), so the benzene will be volatile, so chemical balance will be broken, and toward the direction of benzene formation reaction. Benzene vapour is
cooled and condensed, so that you can collect benzene.
|
|
|
ScienceHideout
Hazard to Others
Posts: 391
Registered: 12-3-2011
Location: In the Source
Member Is Offline
Mood: High Spin
|
|
My Recent Attempts:
Hi guys!
This week I tried three different possible routes to benzene. They all failed, but are worth noting.
Firstly, I know some of you are bound to think that I have been sniffing too many of my reagents. I promise you that I am sane. The most likely reason
why you would be led to such a hypothesis is because of the fact that I am using rather odd reagents to do such reactions. The reason is that I prefer
not working with mixtures of ungodly hot hydroxides and benzoic acid... just something I prefer to stay away from. So here, as follows, are my recent
experiments, and also one of my ideas 
1) REDUCTIVE DE-AMINATION A couple days ago I wanted to try to make it from aniline. I took about 2 mL of aniline and added to it a stoiciometrically
correct amount of HCl to produce a required amount of HNO2 when a nitrite is added in situ. I then made the nitrite solution and added it to
get a diazonium salt. After the salt was made, I tried reducing it with a HUGE excess of NaBH4. Borohydrides have four working hydrides, so I figured
a fourth of an equivalent should work, but I doubled it and chose to do a 2:1 molar ratio of diazonium salt to NaBH4. As I added it, it made a fizzing
sound but no other change was noted. I got, instead, a black tar in the reaction vial and it didn't smell like benzene, so I disposed of it. The tar
was soluble in most polar organic solvents, though.
What went wrong?: I considered for a moment that the BH4- might have been reacting with the water instead of the diazonium, but I know that
borohydrides react with water slower than it reacts with other chemicals. Maybe a hydride just won't react well with this, I can't find a mechanism.
Plus, see my note for the next step.
2) In this experiment, conducted the first steps very similarly (on a somewhat larger scale, .1 mol) but I tried warming the diazonium salt with EtOH
instead of borohydride. I noticed the tar forming again, but in addition I saw many more bubbles than the previous reaction, so I thought it was
working. Cool oily patterns were forming on the surface of the liquid. I dumped it in a sep funnel after it was done bubbling, but saw no separation
like I expected. Instead, the crappy oil came out dropwise and made clean-up a disaster.
What went wrong?: I have no idea. It is either me or my reagents. The bottle of aniline I have is VERY old and has a sort of brown tinge... It also
doesn't smell as pungently as I imagined it would (I inherited this chemical, it came from Merck originally. Must be about 60+ years old. Is there any
way to test it and see if it is still good?
3) ZINC REDUCTION. Here I took .05 mol of phenol and melted it in a 5 mL reaction vial. I added .05 mol of zinc and attached a hickmann still head
with a condenser at the top. I ramped up the heat until it started boiling and collected the distillate. I stopped the distillation just before it ran
dry. I then took it apart and got the distillate. It crystallized. I just distilled the phenol. Not what I was after, but if anyone wants the purest
phenol in town you are more than welcome to come to my house and scrape it out of the head!
What went wrong?: Maybe I should've refluxed it? I really don't know. A couple sources say it should've worked
WHAT'S NEXT? How about BENZENE from DOLLAR STORE CHEMICALS!
All you need is some para-dichlorobenzene mothballs, magnesium, ether from engine starting fluid and water! Tell me if you guys think it will work, I
attached the mechanism, but I am very terrible at grignard-type radical mechanisms so please let give me your critiques!
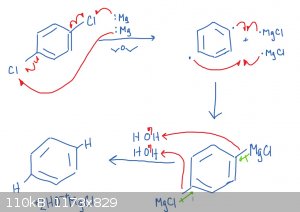
hey, if you are reading this, I can't U2U, but you are always welcome to send me an email!
|
|
|
Morkva
Harmless
Posts: 17
Registered: 6-11-2014
Member Is Offline
Mood: No Mood
|
|
1. Aniline.
If there was an excess of aniline, could it be diazo tars?
Also, copper might help. The N2 needs to come off somehow: copper salt gives an electron and the nitrogen leaves the aryl radical. If you have copper
salt and borohydride in solution I do believe there is at least transient Cu (I) hydride, which may be your key to success, if the issue was not just
separation. Traditionally hypophosphite was used for this kind of reduction, also by a radical mechanism. This kind of radical chemistry is very
interesting to search for and learn about.
2. Phenol. Zinc dust? Maybe if you ran the phenol vapors through a heated tube filled with zinc dust. Nobody can blame for not feeling like doing such
a thing.
3. PDCB
I shall probably eat my words but you may not have to separately form the Grignard reagent or even use ether. Depending on the fondness you might get
away with magnesium metal and alcohol. At least, I saw something like that with calcium metal "arene dechlorination", but then why would they use
calcium? It is also probably possible to take off only one chlorine.
http://archneur.jamanetwork.com/article.aspx?articleid=17887...
In the Octopuses garden...
[Edited on 16-10-2015 by Morkva]
http://www.carrotmuseum.co.uk/falcarinol.html
“Science is the ultimate pornography, analytic activity whose main aim is to isolate objects or events from their contexts in time and space. This
obsession with the specific activity of quantified functions is what science shares with pornography.”
- J.G. Ballard, The Atrocity Exhibition
|
|
|
ScienceHideout
Hazard to Others
Posts: 391
Registered: 12-3-2011
Location: In the Source
Member Is Offline
Mood: High Spin
|
|
Thanks for the response, Morkva!
1) The copper sounds like it would be a good idea, I am just confused as to how I can get those ions in there without introducing any Br- or Cl- that
would cause just a normal sandmeyer reaction. I find it interesting that you bring up the hypophosphite, because every source online uses phosphorous
acid to do the reaction, which is very hard to obtain. I was unaware that it involves a radical mechanism, though. I thought it would be polar.
Perhaps this is why I couldn't figure out how to draw the darn thing 
2) I found many sources that describe passing vapors over red hot zinc dust, but a couple more questionable ones about plain distillation over zinc.
Chemists are lazy; I just had to try the easier (and more practical) of the two! I assume that this is some sort of radical reaction, as well. Maybe
the transition state is too high in energy? In which case, maybe an added catalyst would work.
3) Thanks for the [somewhat comical] link! I will study this! What you said, though, about not having to form grignard reagents in ether or anything
though seems so... simple! I would've thought if it was that easy, others would've already done it...
hey, if you are reading this, I can't U2U, but you are always welcome to send me an email!
|
|
|
Morkva
Harmless
Posts: 17
Registered: 6-11-2014
Member Is Offline
Mood: No Mood
|
|
https://archive.org/stream/aromaticdiazocom031270mbp/aromati...
Look at p. 271 and on, it's about equidistant from the top.
On p. 282 it claims copper hydride works to halogenated by forming cuprous halides, so you're right, you would have to separate the ions and I do not
see how either.
http://www.researchgate.net/profile/Cristian_Simion2/publica...
Phenyl chloride is claimed to be formed quantitatively, and was not reduced further, from dichlorobenzene. Ethanol was the most effective alcohol they
tried. I have no idea about magnesium, except that is too an alkaline earth metal...
If CuH is still interesting to anyone.
http://pubs.acs.org/doi/pdf/10.1021/ic5027009
Open access article on the surface structure of copper hydride produced by different methods. One of these is by Cu(ii) and BH4 in aqua, which forms a
coffee colored precipitate different in color from Würtz's old fashioned CuH which made with hypo phosphorous acid and is red.
Apparently copper hydride can decompose explosively when dried. It also decomposes slowly when wet.
http://journals.iucr.org/b/services/forthcoming.html
Structure and spectroscopy of CuH prepared via borohydride reduction
Elliot L. Bennett, Thomas Wilson, Patrick J. Murphy, Keith Refson, Alex C. Hannon, Silvia Imberti, Samantha K. Callear, Gregory A. Chass and Stewart
F. Parker
We show by a combination of diffraction and spectroscopic methods that CuH produced by borohydride reduction of a CuII salt consists of a core of CuH
with the Wurtzite structure and a shell of water. We also demonstrate that the shell is exchangeable for ethanol.
[Edited on 17-10-2015 by Morkva]
http://www.carrotmuseum.co.uk/falcarinol.html
“Science is the ultimate pornography, analytic activity whose main aim is to isolate objects or events from their contexts in time and space. This
obsession with the specific activity of quantified functions is what science shares with pornography.”
- J.G. Ballard, The Atrocity Exhibition
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
It would not have been unwise to purify the aniline before doing anything with it; of course you know that at the diazo stage, sodium stannite (NaOH
and SnCl2) could have substituted for NaBH4, or the direction could have branched off towards phenylhydrazine with sulfite...which gives benzene with
CuSO4.
|
|
|
clearly_not_atara
International Hazard
Posts: 2834
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
http://en.wikipedia.org/wiki/Benzene_in_soft_drinks
If this reaction occurs in cold soda cans it's probably much faster at high temperatures with higher catalyst loading (more Fe2+/Cu2+ and more
ascorbate). I'm pretty sure that if you can't get benzoate for some reason your next best bet is to oxidize toluene or benzyl alcohol.
Dichlorobenzene will be quite slow to eliminate with magnesium, and probably won't react with zinc or lithium. It could be done by adding
dichlorobenzene to a solution containing ethylmagnesium bromide. In any case it sounds hard :/
Terephthalic acid should decarboxylate as easily as benzoic acid and can be obtained from PETE.
[Edited on 15-11-2015 by clearly_not_atara]
|
|
|
Heavy Walter
Hazard to Others
Posts: 127
Registered: 17-12-2015
Location: Argentina
Member Is Offline
Mood: No Mood
|
|
Hi
Just joined the forum, I learn some people was interested in benzene synthesis.
I experimented with catalyst and acetylene, trimerizing it in order to prepare deuterated benzene. Reaction was quantitative and everything proceeded
as expected, except the small acetylene generator I built, because gas generation was too violent. So I decided to freeze the D2O, landing a small
chunk of calcium carbide on it, closing the generator and then I was able to control the pressure on it opening the way to a storage balloon.
The basic circuit is as follows: gas generator (like a calcimeter), three cold traps (ice, ice & acetone, liquid nitrogen), a storage balloon, one
reactor with the catalyst and a cold finger were the C6D6 was collected while the catalyst was heated.
All the line was evacuated with rough pump and diffusion pump to 10-6 torr.
Regards!
|
|
|
brubei
Hazard to Others
Posts: 188
Registered: 8-3-2015
Location: France
Member Is Offline
Mood: No Mood
|
|
Thx Walter ! That sound very efficient.
For those who tried with diazonium, hypophosphorus acid was the right reducing agent. It seems to be the classical method for reducing aromatic amines
http://chemwiki.ucdavis.edu/Core/Organic_Chemistry/Amines/Re...
One would probably try with other H• radical donor
[Edited on 14-3-2016 by brubei]
[Edited on 14-3-2016 by brubei]
|
|
|
vin123
Harmless
Posts: 2
Registered: 13-3-2016
Member Is Offline
Mood: No Mood
|
|
Depending on the purpose an amount of your preparation of benzene (solvent or building block), in the latter case you may try arenediazonium salt
reduction (starting from aniline). It is possible to trap them with tetrafluoroborate salts to become more stable but I would use them in situ. But be
very carefull when scaling up. For solvent use I would try something else. Moreover, their are many replacements for benzene as a solvent, why using
it if its not available?
|
|
|
Texium
|
Thread Pruned
24-7-2016 at 18:41 |
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Rain today, so tried out the benzene synthesis by dry distillation of 50g sodium benzoate & 14g sodium hydroxide.
(Benzene is the last item required to attempt Magpie's Congo Red synthesis)
Initially a 'golden syrup' tin was used as the reaction vessel, with the remaining distillation glassware (the rest was in the wash).

Using just a small spirit burner, this actually worked (kind of) and gave a tiny, yet totally clear product which smelt distinctly like i always
thought benzene smelled.

A blowtorch was then used to raise the temperature and speed up the reaction. Perhaps doing that caused the next distillate to become orange ?
Unfortunately the metal tin seal began to leak, also the lid, which led to to small benzene fire which was immediately extinguished.
Undeterred, the contents of the tin were extracted and around half was loaded into a 100ml RBF. They're the cheaper to replace than the 250ml+ sizes.

Again the spirit burner was used, this time with aluminium foil for insulation.
The product came over after 8 minutes at a rate of 1 drop every 3 seconds.
The gas escaping from the pot appeared as 'threads' in the condenser.

The result of this distillation was a yellow/orange liquid which clearly has two layers.

The rest of the reagents will be distilled soon, the products combined, then re-distilled to work out the yield.
[Edited on 26-11-2016 by aga]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
This all went a bit wrong and destroyed a 100ml and a 250ml RBF on cooling, as expected.
All of the available sodium benzoate was used (~80g) resulting in about 37ml of orange liquid, still to be re-distilled to get any actual product.
The spread of heat appears to be vitally important.
In an RBF one can see the area affected by the flame becoming black and bubbling, while the rest of the white powder remains unaffected.
With a naked flame (spirit burner) this can be moved to target an unaffected area, which seemed to squeeze out a brief run of more drops of distillate
each time the flame was moved.
A steel conical flask with a huge bottom surface area might work, so that the powder remains in a very thin layer and gets heated enough for the
reaction to complete.
Maybe a steel reactor where an inert atmosphere is super-hot and the powder gets fed in somehow, while allowing the benzene gas to escape.
Edit:
The products were redistilled and a cloudy liquid came over between 71 C and 75 C, the orange stuff stayed in the boiling pot.
Yield at this point is 24.1g = 56.7%
Probably needs drying so that % will reduce 
[Edited on 28-11-2016 by aga]
|
|
|
sulfuric acid is the king
Hazard to Self
Posts: 94
Registered: 11-1-2017
Member Is Offline
|
|
What about coal pyrolisis?...
|
|
|
macckone
Dispenser of practical lab wisdom
Posts: 2171
Registered: 1-3-2013
Location: Over a mile high
Member Is Offline
Mood: Electrical
|
|
Coal pyrolysis is the traditional method but it requires
huge amounts to coal.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
| Quote: Originally posted by aga | This all went a bit wrong and destroyed a 100ml and a 250ml RBF on cooling, as expected.
All of the available sodium benzoate was used (~80g) resulting in about 37ml of orange liquid, still to be re-distilled to get any actual product.
The spread of heat appears to be vitally important.
In an RBF one can see the area affected by the flame becoming black and bubbling, while the rest of the white powder remains unaffected.
With a naked flame (spirit burner) this can be moved to target an unaffected area, which seemed to squeeze out a brief run of more drops of distillate
each time the flame was moved.
A steel conical flask with a huge bottom surface area might work, so that the powder remains in a very thin layer and gets heated enough for the
reaction to complete.
Maybe a steel reactor where an inert atmosphere is super-hot and the powder gets fed in somehow, while allowing the benzene gas to escape.
Edit:
The products were redistilled and a cloudy liquid came over between 71 C and 75 C, the orange stuff stayed in the boiling pot.
Yield at this point is 24.1g = 56.7%
Probably needs drying so that % will reduce
[Edited on 28-11-2016 by aga] |
I made my benzene in steel solvent cans. I punched a hole in the top and attached a brass compression fitting, which I connected to a copper tube that
ran through a thermometer adapter into a condenser. I loaded a powdered mixture of sodium hydroxide and sodium benzoate into the cans, and heated I
heated the cans over a propane stove. This resulted in an orange distillate, which was washed, dried, and fractionated. Each can is good for several
runs.
The yield was far from quantitative, but the reaction is easy to do, and the reactants are cheap and readily available. I was very careful not to
breathe any benzene; I'm not really sure how severe a carcinogen it is, but it has been proven to actually cause certain cancers.
[Edited on 27-2-2017 by JJay]
|
|
|
The jersey rebel
Hazard to Self
Posts: 76
Registered: 27-5-2016
Location: Jersey Fresh
Member Is Offline
Mood: dealing with excessive change
|
|
The methods I am aware of that are accessible to the amateur are demethylation of methylbenzenes, oxidation of toluene to benzoic acid and
decarboxylate the acid over copper chromite or sodium hydroxide, if acetylene is readily available it can be catalytically cyclotrimerized to form
benzene with a cyclotetramer side reaction, Decarboxylation of terephthalic acid over a catalyst such as basic copper carbonate or copper chromite,
acetone trimerization to form the methylated threefold ketone of benzene, and deoxygenation of phenol with powdered zinc. There are numerous other
methods though the feasibility is entirely contingent on where you live and your equipment/experience. These methods are for those who happened to
live on the east cost of the US. Accessibility will differ from country to country and even on the state level.
|
|
|
plastics
Hazard to Others
Posts: 141
Registered: 6-11-2009
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by JJay | | Quote: Originally posted by aga | This all went a bit wrong and destroyed a 100ml and a 250ml RBF on cooling, as expected.
All of the available sodium benzoate was used (~80g) resulting in about 37ml of orange liquid, still to be re-distilled to get any actual product.
The spread of heat appears to be vitally important.
In an RBF one can see the area affected by the flame becoming black and bubbling, while the rest of the white powder remains unaffected.
With a naked flame (spirit burner) this can be moved to target an unaffected area, which seemed to squeeze out a brief run of more drops of distillate
each time the flame was moved.
A steel conical flask with a huge bottom surface area might work, so that the powder remains in a very thin layer and gets heated enough for the
reaction to complete.
Maybe a steel reactor where an inert atmosphere is super-hot and the powder gets fed in somehow, while allowing the benzene gas to escape.
Edit:
The products were redistilled and a cloudy liquid came over between 71 C and 75 C, the orange stuff stayed in the boiling pot.
Yield at this point is 24.1g = 56.7%
Probably needs drying so that % will reduce
[Edited on 28-11-2016 by aga] |
I made my benzene in steel solvent cans. I punched a hole in the top and attached a brass compression fitting, which I connected to a copper tube that
ran through a thermometer adapter into a condenser. I loaded a powdered mixture of sodium hydroxide and sodium benzoate into the cans, and heated I
heated the cans over a propane stove. This resulted in an orange distillate, which was washed, dried, and fractionated. Each can is good for several
runs.
The yield was far from quantitative, but the reaction is easy to do, and the reactants are cheap and readily available. I was very careful not to
breathe any benzene; I'm not really sure how severe a carcinogen it is, but it has been proven to actually cause certain cancers.
[Edited on 27-2-2017 by JJay] |
Exactly the way I did it. Stainless biscuit barrel, tank connector and some 15/22mm copper pipe plus fittings. Yield not great but better than
nothing. I got the same orange distillate which I then redistilled in a glass setup

|
|
|
Texium
|
Thread Pruned
15-6-2017 at 10:17 |
| Pages:
1
..
10
11
12
13
14 |









