| Pages:
1
..
15
16
17
18
19
..
33 |
Darkstar
Hazard to Others

Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Quote: Originally posted by aga  | | Are the OC answers open to everyone, and should be U2U'd to you Darkstar, same as for the QM answers to blogfast ? |
Those questions were primarily intended for you personally. You can just post your answers here in the thread if you want. I'll post the correct
answers afterwards. This way, anyone who's been quietly following this thread can formulate their own answers and then check to see if they were right
or wrong after I post the correct ones.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
I'm honoured !
OK. Here goes.
OC Q1
Step 1
The OH- has one electron too many.
The ethyl acetate seems to have all of it's atoms quite happy, with no free electrons.
The Carbon at the second position from the left will be having the electron density pulled away from it towards the more electronegative Oxygen
(orbital probability density closer to the O than the C) effectively making it relatively electron-poor.
That should make the O of the OH- the nucleophile and the 2nd C of the ethyl acetate the electrophile.
Step 3
The O on the ethanol has the extra electron so must be the nucleophile.
The right-hand H on the sodium acetate (where's the Na ?) couldn't give a shit, as it has it's orbital full and happy, so the
electrophile must be where the electron ends up, which is on the Right-most O of the sodium acetate.
OC Q2
Hmm. Will give it a go.
Step 1
The OH- bonds with the 2nd C giving it a three-O problem, causing the pi bond to the double bonded O to be disrupted.
This leaves the orbitals all in a mess, and an electron homeless.
Step 2
The homeless electron ends up with the right-hand O for no good reason i can think of, other than that it has a spare p orbital for it to half-occupy.
Step 3.
Being less electronegative due to this extra electron, the O on the ethanol structure is ejected, along with the rest of the ethanol structure.
The spare electron is sucked up by the right-most O on the ethyl acetate (via the adjacent H) for no good reason that i can think of.
Extra Credit at the B&D Education Store
Erm, dunno.
After Step #1 there's already an electron excess, so further excess electrons can't have much effect ?
Needs some H+ hanging about to knock out that extra electron ?
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Might be an idea to go U2U on this with a time limit, same as the QM Q/A thing.
The thread might fill up with junk otherwise.
Appologies if what i recently posted also constitutes Junk.
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
I'm just going to post the answers now so we can move on. The real purpose of this quiz was to gauge where you are at the moment so I can determine if
there are any concepts that we still need to work on before we get a bit more advanced. I'm going to go ahead and post the correct answers below and
then personally address your answers in a separate post. The correct answers are:
| Quote: | Answers to OC Quiz #1
Question 1 - Identify the electrophile and nulcleophile in steps one and three of the mechanism.
Correct Answer: In step one, the nucleophile is the hydroxide ion and the electrophile is the carbon
of the ester group. In step three, the nucleophile is the ethoxide ion and the electrophile is the hydrogen of the carboxylic acid group.
Question 2 - Briefly explain what is going on in each step of the mechanism.
Correct Answer: In the first step, the nucleophilic hydroxide ion attacks the electrophilic carbon
of the ester group by donating one of its lone pairs to the electron-deficient carbon center. This breaks the pi-bond between carbon and the carbonyl
oxygen, creating the negatively-charged tetrahedral intermediate seen in step two.
In the second step, a lone pair comes down from the top oxygen to form a new pi-bond with the electron-deficient carbon, simultaneously breaking the
sigma bond between carbon and the ethoxy group and allowing the latter to leave as an ethoxide ion.
In the last step, the ethoxide ion grabs the acidic proton off of acetic acid, forming an acetate ion and a neutral ethanol molecule.
Extra Credit Question - Explain why the reaction is virtually irreversible under basic conditions.
Correct Answer: The second step of the mechanism is reversible because it is possible for the
pi-bond formation to end up breaking the bond between carbon and the hydroxyl group instead, allowing it to leave as a hydroxide ion. This gives back
the original ethyl acetate molecule and the reaction starts all over again. Similarly, step three is also reversible because there are two possible
electrophiles for the nucleophilic ethoxide ion to attack: the hydrogen and carbon of the carboxylic acid group, both of which are electron-deficient.
If ethoxide attacks the carbon, the pi-bond breaks once again to return us back to the charged tetrahedral intermediate in step two. On the other
hand, if ethoxide attacks the proton, a much weaker nucleophile is formed--ethanol. And because there's now a negative charge on the acetate ion, it
is no longer electrophilic enough to be attacked by most nucleophiles--especially not one as weak as ethanol--and is protected by the electrostatic
repulsion caused by the negative charge. Thus as long as the conditions are moderately basic, the acetate ion will remain deprotonated and unable to
undergo further nucleophilic attack. |
[Edited on 11-5-2015 by Darkstar]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Now that I've given out the official answers, I would like to make a few comments regarding your answers:
| Quote: Originally posted by aga | The Carbon at the second position from the left will be having the electron density pulled away from it towards the more electronegative Oxygen
(orbital probability density closer to the O than the C) effectively making it relatively electron-poor.
That should make the O of the OH- the nucleophile and the 2nd C of the ethyl acetate the electrophile. |
Correct!
| Quote: Originally posted by aga | | The O on the ethanol has the extra electron so must be the nucleophile.The right-hand H on the sodium acetate (where's the Na ?) couldn't give a shit,
as it has it's orbital full and happy, so the electrophile must be where the electron ends up, which is on the Right-most O of the sodium acetate.
|
The first part is correct about ethoxide being the nucleophile; however, the electrophile in the third step is the hydrogen! Just because it has a
full orbital doesn't necessarily mean that it's happy. Like the carbon in the first step, hydrogen also has a fairly strong partial positive charge on
it due to oxygen's electronegativity, making it a prime target for nucleophiles. Not to mention that the acetate ion's negative charge isn't actually
localized on oxygen like it appears above. Because of resonance, the negative charge on oxygen that results from removing that proton is relatively
weak. This makes it easier to remove the hydrogen because it is less strongly attracted back to the oxygen.
I'll do a brief lesson soon on what makes certain hydrogens more "acidic" (easily removable) than others, but for now just think of it as having to do
with the degree that the electron density is being pulled away from them. The closer the electron density is to the hydrogen, the stronger it will be
attracted to it and the harder it will be to pull away. On the other hand, when the electrons are being pulled far away from hydrogen, it becomes
easier to remove because its attraction to those electrons (and their attraction back) is weaker.
In the case of carboxylic acids (like acetic acid), the negative charge that ends up on oxygen when the proton leaves is rather weak due to electron
delocalization. Because of this, the hydrogen is quite easily removed because it is less attracted back as it leaves. Which is also why ethoxide is a
MUCH stronger base than acetate, as the negative charge on ethoxide is more or less completely localized on oxygen. And because it's attached to an
electron-donating carbon chain, ethoxide's negative charge ends up being extremely strong, even stronger than hydroxide's. This is what's
known as a superbase. These kinds of bases can't exist in water because they instantly grab a proton off of water to create a weaker hydroxide ion that isn't
strong enough to grab its proton back.
Also, as far as sodium and acetate ions are concerned, there aren't any shown in the mechanism. In the last step, acetic acid is deprotonated to give
the acetate ion, which is only shown as one of the products in the overall reaction above the mechanism. I didn't bother to show the sodium ions in
the mechanism, but they are always around, floating in solution. If the solution were evaporated, the oppositely-charged sodium and acetate ions would
remain and counter one another, giving an ionic salt (sodium acetate).
| Quote: Originally posted by aga | | The homeless electron ends up with the right-hand O for no good reason i can think of, other than that it has a spare p orbital for it to half-occupy.
|
As I mentioned in the answer section, it can end up with either oxygen (hydroxide or ethoxide). The only difference is that, when it ends up with
ethoxide, in the event that it grabs a proton to give an acetate ion and an ethanol molecule, the reaction is over.
| Quote: Originally posted by aga | | After Step #1 there's already an electron excess, so further excess electrons can't have much effect ? |
Remember that there's really a partial negative charge (-1/3) on the carbon of the acetate ion due to electron delocalization. Electrostatic repulsion
would prevent any further nucleophilic attacks on carbon.
[Edited on 11-5-2015 by Darkstar]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Oh !
So the whole resonance thing is imagining (even calculating!) where the electron densities are on the molecule, especially the active bits, and the
relative electrostatic charges that implies ?
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga | Oh !
So the whole resonance thing is imagining (even calculating!) where the electron densities are on the molecule, especially the active bits, and the
relative electrostatic charges that implies ?
|
Basically, when it comes to reaction mechanisms, the whole point of QChemistry is working out the electron probability densities of molecules. These
tell us where electrophilic (Lewis acids) or nucleophilic (Lewis bases) transformative attacks can take place: REACTIONS!
PS: don't forget Q 6!
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Cool !
So imagining the electron jelly bubble around each atom (and how thick/dense it is) is actually Useful !
Knew that would come in handy one day.
Pushing my ignorance a bit further ...
A Carbon atom with 4 bonds should be happy.
What we're discussing is that it's not always the case.
An adjacent bonded Oxygen is being greedy with the electrons, and the Carbon wants more, so is receptive to suitors bearing negatively charged Gifts ?
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga |
A Carbon atom with 4 bonds should be happy.
What we're discussing is that it's not always the case.
An adjacent bonded Oxygen is being greedy with the electrons, and the Carbon wants more, so is receptive to suitors bearing negatively charged Gifts ?
|
Let's say that there are often energetically more favourable arrangements:
O
||
-C- (carbonyl) and -CH<sub>2</sub>- (methylene) both have 4 bonds on the C atom but the formation of a carbonyl group is more
exoenergetic than the formation of a methylene group.
Now does this mean that compounds that contain methylene groups are all queuing up to be 'carbonylised'? No. As it happens that conversion has a high
activation energy, so does not really happen spontaneously: thermodynamics does not equal kinetics!
[Edited on 8-11-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Aha !
"Thermodynamics makes no pronouncement about Kinetics" leaps to mind.
AKA "Just cos it is possible does not make it happen"
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga | Aha !
"Thermodynamics makes no pronouncement about Kinetics" leaps to mind.
AKA "Just cos it is possible does not make it happen" |
Exactly. Think butane lighter: no spark, no flame and yet Free Energy Change of butane combustion is plenty negative...
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
OK. Exhausted my Butane lighter testing that out.
Can i bill the B&D Education Store for the replacement butane ?
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
QQuiz Questions for next week: U2U me the answers!
Question 7: (for 10 points)
Estimate the Enthalpy of Formation (at 298 K) of fluoromethane (CH<sub>3</sub>F), from bond Enthalpies. Show your calculation.
Question 8: (for 20 points)
When 1-chloropropane is refluxed with a small stoichiometric excess of strong NaOH solution, 1-propanol and a solution of NaCl is obtained according
to the following overall reaction equation:
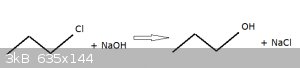
Propose a simple reaction mechanism (use curly arrows to show the movements of electrons).
[Edited on 8-11-2015 by blogfast25]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
| Quote: Originally posted by aga | Oh !
So the whole resonance thing is imagining (even calculating!) where the electron densities are on the molecule, especially the active bits, and the
relative electrostatic charges that implies ? |
There's a lot to resonance. It can help explain anything from why one molecule is more stable than another, to why certain reactions work with some
molecules but fail with others. As I've said, having a good grasp on the idea behind resonance is extremely important in OC. There's a reason I keep
stressing this concept and even dedicated an entire post to showing examples of resonance structures! So just to recap, in the previous quiz, we used resonance to explain:
a) why the proton on acetic acid's hydroxyl group is far easier to remove than the proton on ethanol's hydroxyl group; and
b) why the carbon of the acetate ion is no longer electrophilic enough to undergo further nucleophilic attack.
In the former (a), the negative charge on acetic acid's conjugate base (the acetate ion) ends up getting distributed over three different atoms due to
electron delocalization, making it significantly weaker than the localized negative charge on ethanol's conjugate base (the ethoxide ion). Because of
this, it is much easier to pull a hydrogen off of acetic acid than it is to pull one off of ethanol, as the attraction of the positively-charged
hydrogen back to the negatively-charged oxygen is far weaker in acetic acid. Thus acetic acid is said to be a stronger acid than
ethanol, while the ethoxide ion is said to be a stronger base than the acetate ion. In the latter (b), the partial-negative charge
(-1/3) on the carboxyl-group carbon of the acetate ion protects it from any further nucleophilic attacks via electrostatic repulsion, making the
reaction irreversible as long as the conditions remain basic enough to keep the acetate ion deprotonated.
To help further illustrate the impact that resonance can have on OC reactions, in my next post, I'm going to show you a rather interesting reaction
that I have come to believe ONLY works due to resonance. I will contrast this reaction with another reaction that, due to a lack of resonance
stabilization, ends up proceeding through a completely different mechanism, despite the fact that both reactions use similar substrates under the same
conditions.
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
| Quote: Originally posted by Darkstar | | To help further illustrate the impact that resonance can have on OC reactions, in my next post, I'm going to show you a rather interesting reaction
that I have come to believe ONLY works due to resonance. I will contrast this reaction with another reaction that, due to a lack of resonance
stabilization, ends up proceeding through a completely different mechanism, despite the fact that both reactions use similar substrates under the same
conditions. |
So a couple of months ago I made a post in an OC thread called HI+P reduction mechanism--a thread about the mechanism for the reduction of benzyl alcohols using hydroiodic acid (HI) and red phosphorous. If
this sort of reaction sounds familiar to you, it's probably because it's commonly used by clandestine chemists to reduce l-ephedrine and
d-pseudoephedrine (both benzyl alcohols) to d-methamphetamine (we'll talk about what the d- and l- prefixes mean when we get to chirality), and is the
reason that iodine and red phosphorous are so closely watched in places like the US. This type of reduction is extremely well-known and was first
discovered more than 100 years ago; however, the exact nature of its mechanism remains a mystery to this day, and there's still no unanimous consensus
as to how it actually works. In the thread mentioned above, I proposed a mechanism for it (which you will see below) that is based on the various
conclusions that I have found in the literature over the years.
But before I show you the reaction between HI and benzyl alcohols, I want to show you what a typical reaction between HI and an alcohol looks
like. Since the benzyl alcohol in the example reaction is 1-phenylethanol, I will show you how HI normally reacts with secondary (and tertiary)
alcohols. A secondary alcohol is one where the hydroxyl group is connected to a carbon that is further connected to two other carbons, by the way.
Both of the reactions below involve heating a secondary alcohol with concentrated hydroiodic acid; however, despite being similar alcohols, they give
completely different products. In one reaction, the hydroxyl group gets replaced by an iodine atom, which is what we would expect from treating an
alcohol with a hydrohalic acid. In the other reaction, however, something unique and unexpected happens: the hydroxyl group gets replaced by a
hydrogen atom instead! (what "reduction" means in organic chemistry)
So let's take a look at the usual reaction between secondary alcohols and HI. In this example, HI is reacted with propan-2-ol, also known as
2-propanol, isopropyl alcohol or simply "rubbing alcohol." Here, the hydroxyl group gets replaced with an iodo group via nucleophilic substitution,
producing 2-iodopropane and a water molecule. This type of reaction is called an Sn1 reaction (unimolecular nucleophilic substitution) and
is one of the most common types of reactions in OC. Here's the reaction and mechanism:

In the interest of keeping this post as short as possible, I will just briefly sum up what's happening:
Step 1 - The nucleophilic oxygen of the hydroxyl group donates a lone pair to the electrophilic hydrogen on hydronium. (remember that
HI, when dissolved in water, is really in the form of a bunch of H3O<sup>+</sup> and I<sup>–</sup> ions) This
breaks the old bond on hydronium and creates a new one on isopropyl alcohol, giving a charged oxonium ion.
Step 2 - The oxonium group on isopropyl alcohol takes the electrons with it and leaves as neutral water. This generates a
positively-charged carbon (called a carbocation) at position-2, where the carbon has three bonds and is short an electron.
Step 3 to Step 4 - The nucleophilic iodide ion donates a lone pair to the electrophilic carbon, filling its vacant orbital and
forming a new bond that neutralizes the charges.
This is the usual reaction between secondary alcohols and HI. Seems simple and straightforward enough, right? But when a benzyl alcohol is used in
that same reaction, something strange happens. Like before, the hydroxyl group leaves and gets replaced by iodine (it's arguable whether or not it
actually does, but for now we'll just assume it does); however, the molecule then reacts again and ends up getting reduced by iodide, giving the
reduced alcohol, I2 and water. This reaction is a lot more complex than the ones we've looked at so far and involves radical intermediates,
so don't worry if the mechanism makes absolutely zero sense after Step 2. A radical is when an atom has a single, unpaired valence electron, by the
way. (which typically makes it extremely reactive) The arrows that look like they have a fishhook on the end mean that a single electron is
being moved:

If you want more details regarding this mechanism, just let me know. Basically, the reaction proceeds as follows:
Step 1 - The hydroxyl group is protonated and leaves as water, giving a resonance-stabilized carbocation at the benzylic position.
Step 2 - Iodide attacks the carbocation and forms a new bond.
Step 3 - The weak carbon-iodine bond cleaves homolytically to give a resonance-stabilized benzylic radical and an iodine radical.
Step 4 - The benzylic radical is reduced via SET (single-electron transfer) from a nearby iodide ion to give a negatively-charged
carbanion (essentially the opposite of a carbocation) and a second iodine radical.
Step 5 to Step 6 - The carbanion grabs a proton from hydronium to give the reduced alcohol and the two iodine radicals combine to
give I2.
This is how I believe the reaction works, which is made possible through a combination of resonance-stabilization and iodine's unique properties due
to its size. This mechanism is suggested in the literature and is further supported by the fact that most other alcohols that have a pi-system
directly adjacent to the hydroxyl group get reduced as well, strongly suggesting some sort of resonance-stabilized radical intermediate. The benzylic
radical and iodine radical seen in Step 4 are created via homolytic cleavage of the carbon-iodine bond, which is shown in
Step 3. Homolytic cleavage is where the bond breaks and each atom takes one electron with it. Many times this requires a considerable
amount of energy to do. But because of iodine's size, becoming a radical isn't too difficult for it; however, for a secondary carbon atom, normally it
wouldn't want anything to do with that sort of thing. And as you can see in the reaction with isopropyl alcohol, there's no breakage of the C-I bond
to generate radicals and thus no reduction.
But because of the unique stability of benzylic radicals due to the adjacent aromatic ring, the already-weak carbon-iodine bond becomes even weaker.
(keep in mind that iodine-carbon bonds are already weak bonds to begin with) The benzylic radical is stabilized through resonance like this:

And it is this resonance that makes the bond between the benzylic carbon and the iodine atom extremely easy to break homolytically and the unique
reduction possible. The radical that would be generated in the first reaction with isopropyl alcohol if the C-I bond were cleaved homolytically would
be a lot more unstable due to a lack of resonance, making the radical much more reactive and thus a lot more difficult to create. (still possible,
though, just not as easy. in fact, at high enough temperatures, HI will even fully reduce carboxylic acids!)
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Revisiting the Wave Function
On stuff that doesn’t oscillate, determinism and the particle-wave duality
We’ve come this far in the QM/WM/QC webinar that the time has come to revisit (and hopefully further clarify) one the most fundamental concepts of
this paradigm, the wave function ψ itself. Early on we started with the following definition:

We’ve since worked out the wave functions for a number of simple quantum systems and below we find the schematised wave functions of the ground
state and first two excitations for a single particle in a 1 dimensional finite well (well potential is U<sub>0</sub>, instead of our
usual notation V<sub>0</sub> : :

We’ve also seen that the wave functions ψ and associated energy values E are the eigensolutions of the time independent Schrödinger equation (here
in 1D):
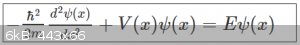
At this point it’s necessary to dispel one of the most profoundly and frequently held misconceptions about Quantum wave functions that new students
of the quantum world often entertain: the wave function as a ‘matter wave’. This pernicious, wrong and stubborn belief stems mainly from poorly
digested interpretations of the de Broglie Hypothesis, skewed readings of the wave-particle duality and teaching analogies involving ‘standing
matter waves’.
Looking at the wave functions of the finite well potential it is indeed very tempting to make the analogy with the standing waves that occur in a
string of a musical instrument, with the ground state the lowest base frequency (that determines the 'pitch' of the note) and the excited states the
so-called harmonics (that determine the 'timbre' of the note).
But nothing could be further removed from the truth: as one physicist famously remarked about quantum wave functions, “nothing oscillates
there!” As we’ll shortly see, it would have been more complete to add, “apart from probabilities!”
In the current and prevailing Born interpretation, the wave function is a probability function and the peaks and throughs probability
amplitudes:

Here P(x) is the probability density distribution of the particle over its domain and the actual probability (of finding
the particle) in an interval [x<sub>1</sub>, x<sub>2</sub>] to find is given by P( x<sub>2</sub>,
x<sub>1</sub>.
(Note: these relationships hold for Real and normalised wave functions)
The term ‘wave function’ is therefore somewhat misleading: unlike in water or sound waves, for ‘quantum waves’ there is no oscillating medium
(water, air or any other elastic medium) and no propagation, so typical of water and sound waves.
Another misconception about QM that needs dispelling is: ‘QM is non-deterministic’, which largely stems from its seemingly probabilistic nature.
But at least at the most basic level one can show this isn’t true.
We saw earlier on that the expectation value < x > can be calculated from a (Real and normalised) wave function as follows:
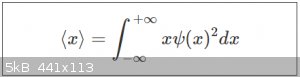
By the expectation value < x > has to be understood the average position of the particle, taken over a very large number of
measurements. For a single particle in an 1D infinite potential well of length L, we found that:
< x > = L/2
If using a detector we measure this value over a very large number of individual observations (or by observing a very large number of identical
systems simultaneously) we always find < x > = L/2. Perfectly deterministic, in other words.
Finally, if quantum wave functions don't describe real waves, where does that leaves us with regards to the particle-wave duality we started this
webinar from? Certainly moving subatomic particles and photons do sometimes behave like waves and sometimes like particles and we don't really have a
good word for that. But to describe how moving particles show wave-like properties like diffraction, modern QM needs not to describe the particle as a
‘matter wave’. Instead the formalism of the particle as a probability wave is sufficient, so there is no contradiction there.
[Edited on 8-11-2015 by blogfast25]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
I think we scared aga away.
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Brief Introduction to Wedge-Dash Notation
When I have a little more time I'll do a proper lesson on chirality. For the time being, I've prepared a mini-lesson on wedge-dash notation. This
will serve as a lead-in to enantiomers by familiarizing any young OC Padawans following this thread (*cough* aga *cough*) with the concept of
representing three-dimensional molecules in only two dimensions.
While drawing molecules using the line-angle formula is undoubtedly the most practical way to draw molecules in OC, the problem is that molecules
aren't actually two-dimensional in real life. For non-chiral molecules (we will cover chirality soon) this isn't really a problem; however, for
molecules with one or more chiral centers, we need a way to represent which direction certain bonds are oriented in three-dimensional space because it
makes a difference. With that said, I present to you the wedge-dash notation:

As you can see, the normal lines are oriented along the plane of the image; the wedged lines are pointing towards the viewer; the dashed lines are
pointing away from the viewer. Compare the wedge-dash drawing of propane above to the ball-and-stick model of propane below:

Notice that the bonds are oriented the exact same way as the bonds in the wedge-dash drawing suggest.
Extra Credit:

In the image above, I have drawn 2-chloropropane in the top box and (1-chloroethyl)benzene in the bottom box. In each case, I have drawn the molecule
two different ways: one with the chloro group pointing towards the viewer, and one with the chloro group pointing away from the viewer.
For extra credit, explain why the two molecules in the top box are the exact same molecule, while the two molecules in the bottom box are not. What
have I changed by adding a phenyl ring?
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Not a chance !
There are a lot of pitchforks and red squiggly lines to absorb in your recent posts, which will take a little time.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Answers to last week's QQuiz questions:
| Quote: Originally posted by blogfast25 |
Question 5: (for 10 points)
Predict the geometrical shape of the following molecules:
a) Xenon difluoride, XeF<sub>2</sub>.
b) The pentafluoride xenonate anion, XeF<sub>5</sub><sup>-</sup>.
Question 6: (for 10 points)
Dinitrogen difluoride (N<sub>2</sub>F<sub>2</sub> has two
isomers, as shown below, left trans-N<sub>2</sub>F<sub>2</sub>, right cis-N<sub>2</sub>F<sub>2</sub>:
Briefly explain why two such isomers exist.
|
A. 5:
a. XeF<sub>2</sub>:
Both bonds are identical. Electrostatic repulsion between the bonding MOs is minimised if the molecule takes on a linear shape: F-Xe-F.
b. XeF<sub>5</sub><sup>-</sup>:
All bonds are identical and the single charge is delocalised. Electrostatic repulsion between the bonding MOs is minimised if the molecule takes on a
trigonal bipyramidal shape as shown here.
A. 6:
Two of three valence electron of each N contribute to the double bond: two p<sub>x</sub> form a σ(pp) MO, two p<sub>y</sub>
form a π(pp) MO, forming a torsionally rigid structure.
The F atoms can then bond to the remaining N valence electron on either side of the double bond. So two different structures, called 'cis' (same side)
and 'trans' (one on each side) arise.
Additional note to A. 6:
Because of the abundance of double C bonds in organic chemistry, cis/trans isomerism is very common in OC, see below for example the structures for
2-butene:

|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Bond Enthalpies Erratum:
In the post on bond Enthalpies the following error was not previously spotted:
| Quote: Originally posted by blogfast25 | Bond Enthalpies:
Estimating reaction Enthalpies from bond Enthalpies:
These values can be used to estimate the reaction enthalpies of simple reactions. Here, mainly for illustrative purposes, the estimated Enthalpy of
formation of ethane:
2 C + 3 H<sub>2</sub> === > C<sub>2</sub>H<sub>6</sub>, ΔH<sub>f</sub><sup>298K</sup>
To find this value write this equation as a sum of ‘sub reactions’ using Hess’ Law:
1) 2 C === > C – C, yields - 368 kJ
2) 3 X [H<sub>2</sub> === > 6 H.], costs 3 X + 435 = + 1305 kJ
3) C – C + 6 H. === > H<sub>3</sub>C – CH<sub>3</sub>, yields 6 X – 414 = - 2484 kJ
Adding up: ΔH<sub>f</sub><sup>298K</sup> = - 368 + 1305 – 2484 = -1547 kJ per mol
C<sub>2</sub>H<sub>6</sub>.
|
Although reaction Enthalpies can certainly be estimated by the method pointed out in the post, where that reaction is a formation reaction
the Standard Enthalpy of Atomisation of Carbon, value +717 kJ/mol, needs also to be taken into account (this would also be true for the estimation of
formation Enthalpies for non-carbon based covalent compounds, where the atomisation Enthalpy of the main element then needs to be accounted for).
In the case of ethane the calculated value of - 1547 kJ/mol needs to be adjusted by adding 2 X + 717 kJ/mol, which gives us a value of - 113 kJ/mol
for the Standard Enthalpy of Formation of ethane. The generally listed value is - 84 kJ/mol.
Apologies.
[Edited on 9-11-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Now i'm confused by the Questions.
Am i answering QM Q 7 with the revised or original methodology ?
Are there any OC Questions i've missed ?
Still a Whopping chunk of OC to re-read and digest some more (he said, hoping for mercy)
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
As per our U2U use the 'revised'<sup>*</sup> method if you can. Scoring will reflect my error, so it should be an easy 10 smackaroons!
* Revised number = original + 717 kJ/mol. Easier than decimalisation!! 
[Edited on 9-11-2015 by blogfast25]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
| Quote: Originally posted by aga | Are there any OC Questions i've missed ?
Still a Whopping chunk of OC to re-read and digest some more (he said, hoping for mercy) |
There's no rush; take your time. As far as OC questions, the only new one is at the bottom of this post. It's just an extra credit question to serve as a lead-in to our upcoming lesson on chirality and enantiomers, so don't worry about
trying to figure it out unless you have time. It's just to get you thinking about visualizing molecules in three dimensions.
The other posts I made were just more on resonance. This post just more or less reiterates what I've already said before, and this post just talks about an interesting reaction between hydroiodic acid and benzyl alcohols (any alcohol with a pi-system directly adjacent to
the hydroxyl group, really) that is likely a result of resonance stabilization. It's mostly just extracurricular reading since it's a bit more
advanced than anything we've talked about up until now. I will say that the post does briefly introduce you to Sn1 reactions,
however, which are extremely common in OC. It would probably be a good idea to at least read up on that part.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Embroidering slightly more on bond Enthalpies:
In the post on bond Enthalpies I indicated that the listed values should be considered as averages, in the sense that the exact value of a
bond Enthalpy depends on its molecular environment. The following link to a *.pdf illustrates that very well (besides also listing a far greater
number of values):
https://labs.chem.ucsb.edu/zakarian/armen/11---bonddissociat...
(the values are bond dissociation enthalpies, so almost always positive)
Here's a sample of values for various C-C bonds in different molecules to illustrate the point of bond Enthalpy variability:

|
|
|
| Pages:
1
..
15
16
17
18
19
..
33 |








