| Pages:
1
..
20
21
22
23
24
..
33 |
greenlight
National Hazard

Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Thanks nitro-genes, is it possible to add the product back into a beaker with 100 grams 98% sulfuric acid and add the rest of the nitrating mix while
watching temp?
Otherwise, I will continue with the third step and see how it goes.
There is no amount specified on your writeup on this thread for the third step so I am assuming that all the dinitroacetaminophen recovered from the
second step is used.
[Edited on 11-9-2015 by greenlight]
Be good, otherwise be good at it 
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Dissolving the precipitate again may be very difficult, you could try dissolving at 10 deg C. again, but do it with half the precipitate you obtained.
There is no amount specified, because unless recrystallized, the DNAc will slowly partly deacetylate from the remaining acid impurities, so I usually
just continue anyway. The deacetylation step is not very critical regarding the amount of sulfuric acid present, 12.5 ml of 96% SA should be enough
for even a 100% yield to deactylate and form the isopicramic sulfate.
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
If I have enough spare time I will try and see if it re-dissolves otherwise I will go straight to the deacetylation and post the results of that too
Thanks..
[Edited on 11-9-2015 by greenlight]
Be good, otherwise be good at it
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
It is my guess you have likely gotten a lot of the DNAc in high yield and not much else. I think you should just proceed to deacetylation with what
is probably DNAc that precipitated earlier than expected possibly due to a low water content and a high yield.
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Okay, I will go straight on to the deacetylation, thanks Nitro and Roscoe for the advice..
Be good, otherwise be good at it
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
@ Greenlight, how did the deactylation work out? Can you take a look at your
bottle of 70% HNO3 btw? Is it w/w or v/v? Did you weigh the HNO3 or used ml? If it is v/v, it would explain why all DNAc precipitated out and no
mononitro may be present at all. Should have asked in advance, just .don't know what is more standard commercially, since I usually distil it myself
and add water to a density of 1.42, which translates to 70% w/w
[Edited on 13-9-2015 by nitro-genes]
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
@Nitro-genes, I am doing the deacetylation today or tomorrow latest and will post results again.
The bottle says 68-70% w/w density 1.42 g/ml and I measured the HNO3 for the dinitration in ml's.
Be good, otherwise be good at it
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Quote: Originally posted by nitro-genes  | Hi Philou, thanks for this synthesis scheme, some interesting compounds, some of them never described before, especially the o,p HNBDF looks like a
very interesting molecule, do you think it would be able to form salts? The synthesis route described looks doable, and apart from the NaN3 (which can
be made OTC though) completely OTC. Pretty cool!
Some things I wondered about :
1 How selective would the chlorination of nitrobenzene be in producing only 3,5 dichloro nitrobenzene? This can probably be found in literature, but I
haven't had the time yet to get a good look.
2. How selective is the Fe/HCl reduction in only reacting with the nitro group? Is there completely no dehalogenation? Does it need a certain strenght
of HCl, or are acidic conditions enough? I'm asking, because probably, mono and dichloro aniline would be hard to separate
3. What would be the best solvent for the diazotization step, since dichloroaniline is not soluble in water, though it seems to be in ethanol. Ethanol
can be used in some cases, but can give some coupling products IIRC.
4. It would be really cool to see how easily the resulting 3,5 dichlorophenol would be able to nitrate further as opposed to 1,3,5 trichlorobenzene.
Do you have a reference for this nitration? Curious about how stable the chloro groups would be for the phenol.
5. the 3,5 dichloro 2,4,6 dinitrophenol would be a very nice precursor to have, the reaction with ethanolic hydrazine may produce a Di N-hydroxy
benzotriazole, which has never been described before I think. If it would be stable that is, N-hydroxy benzotriazoles are notorious for explosions
during synthesis IIRC. Definitely a nice mad science project, nice!
Another question for those who may know more about organic chemistry than me:
Benzoxazolone can be made in high yield, completely OTC. From literature it seems the 6 and 5 positions are most activated, and 6-nitro benzoxazolone
can be made from just 65% HNO3 in high yield by nitration at 0 deg C. Now, could you hydrolyze the oxazolone ring using reflux conditions and dilute
mineral acids, or would the nitro group decompose? If you would get 2-amino 5-nitrophenol and nitrate it further, what would be the outcome? I
couldn't find anything on this, not even for 3-nitrophenol. Suggestions for followup or stability of these compounds? This could be really
interesting, as most of the reactions involved are smooth sailing.
|
1) Selectivity is high because nitrobenzene is strong meta-director. Meta-halo-nitrobenzene is obtained in good yields. Normaly
1,3,5-di-halo-nitrobenzene should be obtainable by further treatment with excess réactants, since addition of a single halo atom will not change much
the meta directing effect of the nitrogroup.
Here is a synthesis of 3-chloro-nitrobenzene from isocyanuric acid trichloride and H2SO4
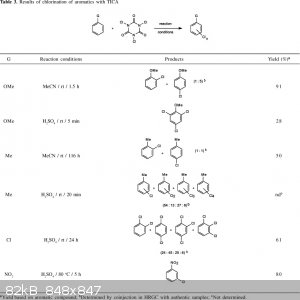
Alternatively one may get acces to 1,3-dihalo-5-nitrobenzene in high yields (>95%) via p-nitro-aniline halogenation -->
2,5-dihalo-4-nitro-aniline and reductive diazotation with ethanol --> 1,3-dihalo-5-nitrobenzene
2) Fe/HCl reduction needs aqueous concentrated HCl (>20%) it is very selective of the NO2 group; so halide atoms won't be
hydrolysed...alternatively one could use Sn/HCl, SnCl2/HCl, Zn/HCl or even Zinin reduction with Na2S/S/NaOH. --> 3,5-dihalo-aniline
3) Water will be fine especially if you use the ability of the aniline to form water soluble salts like Ar-NH2.HCl the later could be reacted with
saturated NaNO2 aqueous solution
Ar-NH2.HCl + NaNO2 --> Ar-NH2.HNO2 + NaCl
Ar-NH2.HNO2 --> Ar-NH-N=O + H2O <==> Ar-N=N-OH (diazonium)
4) 3,5-dihalo-phenol should be nitratable easily like phenol since the halide helps a little the introduction of the nitros in a sympathetic pathway
of the OH. The resulting dihalo-TNP will be stable in acidic media, but prone to dehalogenation (hydrolysis) in basic media.
5) Yes hydrazine may enter the molecule at the place of the halide, just like NH3 would or other amines
--> Methylamine for a tetryl variant (3,5-di-methylnitramino-2,4,6-trinitro-phenol  ). ).
HO-C6(NO2)3(-N(NO2)-CH3)2
Care must be taken because depending on conditions hydrazine may reduce some NO2 groups...
On another thought dihydrazino-TNP would maybe form a mono or disalt of HClO4 or HC(NO2)3...
HO-C6(NO2)3(-NH-NH2)2 . 2 HClO4
HO-C6(NO2)3(-NH-NH2)2 . 2 HC(NO2)3
Also interesting the dihydrazino compound upon action of HNO2 will form the diazo-TNP (I like the coherence of chemistry).
6) About your idea of hydrolysis of benzoxazolone, acidic media should be OK without splitting the NO2.
But the resulting o-amino-phenol must be quite oxydisable by the air...probably forming easily o-quinonic compounds.
H2N-C=C-OH -Ox-> HN=C-C=O + H2O (aromatic ring left aside for clarity)
The later may condense with the non-oxydised product forming a 3 colladed extended aromatic ring...
HN=C-C=O + H2N-C=C-OH --> cyclo(-N=C-C=N-C=C-) + 2 H2O
[Edited on 14-9-2015 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes | Viable route to a possible iso-DDNR? What do you guys think?
Up to the reduction of 5-nitro benzoxazolone to 5-amino benzoxazolone is described in literature, the nitration of the latter isn't, so this is
somewhat of a guess that the 4,6 nitro derivative would result, same for the oxidation/diazotization.
|
The process looks fine up to the last step.
The diazotation will occure easily on the NH2 aside the OH and will form a diazonium phenate; the opposite NH2 between the two NO2 will remain a NH2;
there is no reason for it to become an oxygen (p-quinonoid).
H2N-C=C-OH + HNO2 --> O=N-NH-C=C-OH <==> HO-N=N-C=C-OH
HO-N=N-C=C-OH --> (+)N=N-C=C-O(-) + H2O
Diazonium cations can be stabilized by viccinal (ortho)acidic groups like:
-OH (phenol)
-NH2 (aniline)
-CO2H (carboxyl)
-SO3H (sulfonic)
and maybe
-CH2-NO2 (nitromethyl)
-CH(NO)NO2 (nitroso-nitromethyl)
-CH(NO2)2 (dinitromethyl)
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
thanks for explaining all that philou, it's a lot to think about, I need to let it all sink in before making a thorough reply. You are right about the 5,2-amino 4,6-dinitrophenol, the 5 amino group won't oxidize
or form the diazo, same as for picramide probably. The resutling compound, which might be called 2 -diazo 5-amino, 4-6 dinitrophenol might still be
interesting if it exist and might form nitrate salts or perchlorate salts when diazotized in the corresponding acids using nitrite or even complex
with transition metal salts containing nitrates or perchlorates, although this isn't reported for trinitroaniline itself, so maybe that won't happen.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
5-diazo-styphnic acid ....(exist?)
On the preceding page the 3-diazo-picric acid was documented
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
If the hydroxylamine reaction with styphnic acid would occur similarly as with picric acid, then a 5-amino-styphnic acid would result, which could
possibly be diazotized to 5-diazo-styphnic acid.
No search has yet been done to see if this contemplated compound and resorcinol derivative is known.
Even if it is unstable and one of the 3 nitro groups hydrolyzes easily, the resulting dihydroxy-diazo-dinitrophenol-(1-anhydride) compound would
possibly itself be interesting.
Update: the 5-amino-styphnic acid is known CAS 128585-26-2

Formula: C6H4N4O8
Molecular Weight: 260.11800
Synonyms:
5-amino-2,4,6-trinitro-1,3-benzenediol
5-amino-2,4,6-trinitro-resorcinol
The reported synthesis circa 1928 involves 3,5-dinitroaniline being further nitrated to pentanitroaniline and presumably followed by hydrolysis, which
is far more complicated than the possible reaction of hydroxylamine with styphnic acid.
So the precursor 5-amino-styphnic acid exists and upon diazotization may lead to a nitro-iso-DDNR and / or upon hydrolysis of the third nitro may
provide a hydroxy-iso-DDNR.
These compounds would fit the general template for more powerful derivatives of DDNR as described by the Von Herz 1922 patent GB207563
[Edited on 9/15/2015 by Rosco Bodine]
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Deacetylation finished.
Hour under reflux at 95 Degrees, solution went from yellow to dark red, cooled, transferred to beaker and Ammonia solution added in small portions
until p 4.
Brick red precipitate filtered and drying. One of the fastest compounds I've ever filtered
[Edited on 16-9-2015 by greenlight]
  
Be good, otherwise be good at it
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Nice! looks like a good yield too. When it dries, you'll see it will take on a more orange colour. Filtering is very easy indeed, I suspect they are
agglomerates of small crystals, like Dabney has described. Did you filter the deacteylation mix after cooling btw? Or did you see no precipitates in
the dark red solution at all? Usually there is about 100-200 mg of undissolved dark brown stuff in the filter, I suspect some left over impuritities
from the tablets. What solvent and by what method did you purify the
paracetamol?
Are you planning the diazotization using nitrte or sulfuric acid/nitrate salt/copper? Curious if you get the same results, from the copper method I
got incredibly dense and free flowing pDDNP.
[Edited on 16-9-2015 by nitro-genes]
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Thanks! Yes, I cooled down and then filtered and there was a small amount of brown precipitate in with the solution. I extracted the acetaminophen
using the exact method and amounts from your writeup and I used 95% Ethanol.
I was going to attempt the diazotization with Nitric acid but in your writeup it says that excessive temperatures can cause formation of sensitive
sprengel explosives so I am unsure for my first time.
How bad do you think the dangers are?
Otherwise, I will go with the sulfuric acid/ copper/nitrate salt (probably using KNO3) in a fume hood as I am not keen on any NOX
Also, should i be storing the iso-picramic acid I don't use straight away under distiller water?
[Edited on 16-9-2015 by greenlight]
Be good, otherwise be good at it
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Wouldn't use the 65% nitric acid alone synthesis, it give fairly low yields, presumably since much of the product is oxidized, what precipitates seems
very pure though. Not sure about the risks of explosion, 65% nitric acid contains a fair amount of water, if you want to do the reaction with nitric
acid, you could always use a large excess, say 30 ml for 1 gram of isopicramic, but it seems a terrible waste to me.
The copper method produces very nice yield with diluted sulfuric acid and nitrate salt, I tried both ammonium nitrate and potassium nitrate once, the
potassium nitrate seemed to result in slightly higher yield.
Another thing you could try is a combined nitric acid/copper method, something I haven't tried yet:
You could try to add 1 gram of isopicramic acid to 15 ml 20-30% nitric acid, add the copper beads and keep at a temperature for which gentle
generation of NOx is evident. The extra dilution will eliminate explosion hazards and prevent sudden exotherm, with some tweaking, a concentration of
nitric acid and temperature can probably be found that produces good yields, while releasing a slow and minimal amount of NOx to the surrounding.
Regarding storage..I must admit, I have stored it in a dry and airtight container, away from sunlight, since the stability of the compound in
distilled water or in sunlight is presently unknown. Isopicramic acid doesn't appear a direct explosion hazard, upon flame it first needs to melts and
then steadily burns, leaving a lot of carbon. The potassium salt and sodium salts seem somewhat more energetic and could probably explode when burned
under strong confinment. The ammonium salt is much tamer, that is why the deactylation is performed preferably with ammonia.
[Edited on 16-9-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
WRT "Sprengel composition" that was my observation regarding a real safety concern which does arise particularly in mixtures of concentrated HNO3 in
which is dissolved an energetic fuel, because the mixture itself qualifies as an explosive mixture you don't want to be knocking around carelessly or
incorrectly believing that such a mixture is a benign material. Since there are not any literature descriptions of industrial scale batches being
handled uneventfully, there really is no information about the safety and scale implications for these undocumented experimental processes. Lack of
historical information presents an unknown. which should be kept in mind. Scale is definitely a safety factor and temperature is another safety
factor, and with any experimental lab scale process there should be a situational awareness where risk analysis takes into account what is not known.
When doing experiments with energetic materials a healthy level of caution is just good sense more than paranoia.
What nitro-genes is saying about some dilution with H2O of the concentrated HNO3 does greatly increase the safety compared with the more concentrated
HNO3.
[Edited on 9/16/2015 by Rosco Bodine]
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Thanks guys, I think I will stay away from the Nitric acid alone synthesis, not as good yield, waste of Nitric and more hazards=not worth it.
I will try the copper metal, nitrate salt (KNO3) and SA method first because like Roscoe said, it is good to maintain a level of caution with
energetics and I would rather try something that has already been performed with results for the first diazotization. After that, I will attempt with
copper/diluted nitric acid and compare yields from the two procedures.
Be good, otherwise be good at it
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
It was mainly the heating of the isopicramic acid with fuming HNO3 that would possibly be a safety issue, even though that has been done industrially
with picramic acid to make DDNP, as a Dehn patent process, there is an ignition hazard where the batch is covered with mineral oil to prevent the
spontaneous ignition of the fumes by reaction with oxygen in the air. Igniting a vat of DDNP in fuming HNO3 could become a crater geometry exhibit
where a plant used to be
The use of a lesser concentrated HNO3 would likely be much safer and okay especially something like maybe a 35-40% HNO3 would probably be no issue.
Safety is a kind of compromise where a too mild condition quenches a reaction or prevents good yields, but too harsh conditions may offer no advantage
either, and have disadvantages, so there is found some happy middle ground condition where a process is reasonably optimized. It will work that way
for most syntheses where you develop the best process gradually from a kind of trial and error testing to find what works.
|
|
|
dave321
Harmless
Posts: 45
Registered: 22-11-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by greenlight | Deacetylation finished.
Hour under reflux at 95 Degrees, solution went from yellow to dark red, cooled, transferred to beaker and Ammonia solution added in small portions
until p 4.
Brick red precipitate filtered and drying. One of the fastest compounds I've ever filtered
[Edited on 16-9-2015 by greenlight] |
sorry, meant to quote this post ....
looks like excellent product.
how much 0.88 ammonia did it take to get to ~ph4.
I would imagine quite a bit ?
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Thanks Roscoe, I will use the diluted nitric
@Dave, I was using a dropper to dispense the ammonia solution from a bottle so I am unsure of exact amount.
It was a fair bit though, about 100-150ml.
[Edited on 17-9-2015 by greenlight]
Be good, otherwise be good at it
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The diazotization using the copper and a nitric acid solution, or mixed acid solution of the isopicramic nitrate likely provides a means of
controlling and slowing the speed of the crystallization of the p-DDNP so that larger and better crystals are formed. The p-DDNP will tend to remain
dissolved in the acid as the diazonium nitrate if the acid strength is high and water content is low and the temperature is warm, or in the
alternative the p-DDNP would likely be more inclined to precipitate more quickly as finer crystals as it forms from a more diluted acid. If the
acidity goes too low then the copper will not react as wanted, and I think there is a transition at about 20% HNO3 - 80% H2O where copper starts
reacting differently with HNO3 to produce more NO instead of NO2, but the NO would be oxidized by the air to NO2 and may still cause the diazotization
though reacting more slowly and not as efficiently. So there are some ranges of different acid concentrations and temperature and a volume
relationship that is a factor also that is going to affect crystal development, and it is unknown exactly what is optimum in terms of how many ml of
what composition of acids per gram of isopicramic acid and what temperature and reaction time is optimized.
The conditions described by nitro-genes using a mixture of diluted H2SO4 and a nitrate salt may work somewhat differently from a mixture of H2SO4 and
HNO3 and H2O, and the dilution, optimum H2O content, may be different using neat HNO3 instead of a nitrate salt with diluted H2SO4. If a
diazotization is performed where much of the formed p-DDNP remains dissolved as a diazonium nitrate, it may result in larger crystals if the reaction
mixture is very gradually and slowly diluted with H2O instead of the reaction mixture being crashed onto ice or into a large volume of H2O. Generally
the slower the crystallization the larger the crystals grow, and a crash precipitation by dilution or cooling will result in a microcrystalline or
amorphous powder.
If you use the edit button for the double post earlier you can delete the extra reply post.
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
So using the dilute HNO3 is untested ground on here.
I am going to use nitro-genes method with a nitrate salts to get the feel for the reaction and see how much NO2 fumes should be evolving as well as
how the reaction mixture should look while performing the diazotization. This will give me some DDNP to experiment with initiating secondaries with.
Then I will attempt the diazotization with dilute HNO3/H2O mix first using one of the amounts listed in the above post by Nitro-genes (15-20ml 20-30%
HNO3) probably starting with the more dilute acid first. I'm thinking 25% to avoid that transition were more NO is formed than NO2 that you stated.
So the variables performing a 1 gram batch with isopicramic acid and dilute HNO3 would be:
* Nitric acid concentration
* Nitric acid amount used
* Temperature
* Reaction time
There will be an optimum reaction environment, the task is just finding it
Maybe dripping the final reaction mix from an addition funnel into ice water that is being agitated (safely) would afford more desirable DDNP
crystals.
Only other question is will the dilute HNO3 produce a better yield than the nitrate salt method?
[Edited on 17-9-2015 by greenlight]
Be good, otherwise be good at it
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
my guess would be that the yield from mixtures containing sulfuric acid will be higher, since the pDDNP is insoluble in the reaction mixture as
opposed to warm fairly conc. nitric alone. With strongly diluted nitric, the pDDNP may also precipiate directly as it is formed, alhtough increase in
solubility with increasng temperature is unknown.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
@ greenlight Yes the experimental for the use of copper particularly is uncharted territory and is something that so far as I know is a novel method
for diazotization that can be credited to nitro-genes. It can be difficult to get good crystal development for o-DDNP and efforts to achieve getting
an improved density product have been reported in patents, which are methods very specific to the reaction conditions where a narrow range very
specific reaction window is found to produce good crystals, while any variation of the conditions will result in a lowered quality product. The same
problem applies to p-DDNP of identifying what are the best conditions for diazotization that will produce a desirable crystal form that is dense and
granular and free flowing.
The performance of energetic materials is affected as a general rule by the crystalline form and density. So it is good practice when doing the last
step of the process which provides the end product to write down every small detail. Make careful notes about the reaction conditions so the subtle
differences can be charted, and from a series of experiments it can be learned what are the conditions that produce the best product.
This topic is actually a serious scientific research which is being done and reported here about a very obscure energetic material for which there is
almost no published information. The most extensive collection of information available about p-DDNP is found right here in this discussion thread.
[Edited on 9/17/2015 by Rosco Bodine]
|
|
|
greenlight
National Hazard
Posts: 755
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Can the final product be recrystallized via acetone if the proper crystal size is not obtained.
When I attempt the diazotization with dilute HNO3 instead of nitrate salt I will make sure everything is recorded and hopefully an optimum reaction
condition can be found.
And yes, I have noticed that there is very little information available on DDNP anywhere else on the internet but did not know that SM has the most
extensive collection
Be good, otherwise be good at it
|
|
|
| Pages:
1
..
20
21
22
23
24
..
33 |








