| Pages:
1
2 |
Sauron
International Hazard

Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Preparation of anthranilic acid
This compound, o-aminobenzoic acid, or aniline-2-carboxylic acid, is usually prepared, per Vogel, from phthalimide by treatment with hypobromite or
hypochlorite.
I made some phthalimide a couple years ago by three different methods.
Phthalic anhydride and ammonium hydroxide
Pthalic anhydride and urea
Phthalic anhydride and ammonium carbonate
All of which worked nicely.
But I never managed to get Vogel's procedure to work, for making anthranilic acid.
I have now compared Vogel with the procedure in "Fundamental Processes of Dye Chemistry", q.v. and find Vogel somewhat wanting.
However, even the more detailed and somewhat larger scale procedure in this book, is rather fiddly and at some junctures vague.
I therefore propose an entirely different route.
2-nitrobenzoic acid is a commercial product and not too costly.
Org.Syn. has a procedure for the low pressure (3 atm.) hydrogenation of p-aminobenzoic acid ethyl ester with PtO2 catalyst. Sounds like a job for my
Parr 3911 shaker type hydrogenator. This apparatus was designed by Roger Adams for use with his catalyst, and he happens to be author of this
procedure. The yield is quantitative.
Other reductions are referenced.
[Edited on 30-3-2008 by Sauron]
Attachment: CV1P0240.pdf (129kB)
This file has been downloaded 2917 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I had the idea of reducing ethyl p-nitrobenzoate to benzocaine, using Zn/CaCl2. The yields are said to be moderate, but i think a CTH (not having a
Parr shaker myself  )could work nicely here, though i haven't got any specific
reference to back this up. )could work nicely here, though i haven't got any specific
reference to back this up.
I was thinking of chlorinating p-TsOH, oxydizing the chloro-sulfonic acid at the methyl group with dilute HNO3, removing the sulfonic group as in
the o-chlorobenzoic thread, then esterification and reduction.
The only difference i though was keeping the sulfonic group on during the methyl oxydation, as i would be using dilute HNO3. I don't think any
di-nitration could accur with those conditions, but i prefer staying on the safe side. I guess some displacement of the sulfonic group can happen in
those conditions though.
Just an foolish idea, but if after nitrating TsOH with mixed acids, could diluting the mixture directly oxidize the methyl group (dilute HNO3) AND
remove the sulfonic group (dilute H2SO4)? That a long shot, and could give rise to isomer formation, but it would save alot of time!
one-pot-3-reactions..
Sorry if i'm drifting away.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
There's two Model 3911 sets, at least one of them including the 4833 controller, on offer right now. The asking price is $400 each. I know how to get
25% off that, but I already have this model and its controller. These are in Michigan and are US voltage.
This is the 500 ml model. I paid that much for the 3911, and bouth the controller seperately for almost as much again. So whoever gets these will be
getting a great deal. Almost makes me wish I needed more than one of them. But, I don't, although I do lust after the two liter version.
Anyone interested PM me for details.
Sic gorgeamus a los subjectatus nunc.
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I produced about 50 grams of phthalimide several weeks ago for this synthesis. I planning on try it a soon as a get some more MnO2 for making the
required bromine. Did you use glacial acetic acid in the end to precipitate it? Tomorrow, in ochem lab, I am going to synthesize P-aminobenzoic acid
from p-acetotoluidide. This uses the same precipitation procedure. I want to make some anthranillic acid to produce benzyne which will be used in some
Diels-Alder reactions.
|
|
|
Aubrey
Harmless
Posts: 37
Registered: 16-11-2008
Member Is Offline
Mood: No Mood
|
|
I have also attempted the Dye chemistry synth and am planning to try again this weekend. Has anyone had success using the method described in dye
chemistry?
The part that seems vague to me is the quantity of hypochlorite solution to use. It mentions using 2 mol of NaOH for each mole of NaOCl, but it also
mentions putting in 20cc of NaOH immediately beore use. Do I calculate the quantity of NaOCl based on this total figure? How important is the
accuracy of the amount of NaOCl? For the quantities described I calculaed the amount of NaOCl to use at about 1300ml but i dont feel confident in my
calculations.
I'm also interested to know how important it is to neutralise it at 80 since other sources mention cooling it before neutralising.
It also mentioned (elsewherre) not to use too much HCl because it could destroy the anthranilic, but then it says the filtrate is acidifed with 40cc
of HCl and 12cc glacial.
I'd be very interested to hear if anyone could shed light the above points. If only I had a Par apparatus.
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
O-aminobenzoic acid is a amino acid and has a very narrow isoelectric point. This means that it only precipitates at a certain pH in high yeild. I was
able to obtain about 15g of crude anthranilic acid from this reaction. The neutralization is the tricky part. The reaction mixture is first brought to
about pH 7 with hydrochloric acid and glacial acetic acid is slowly added with stirring until precipitation of the dark colored product is nearly
complete; easy to over shoot. The product is then filtered off and recrystallized from water. The product is tan in color and melts sevral degrees
below the theoretical value.
Amateur NMR spectroscopist
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
One could consider starting from easily available ortho-nitrotoluene.
CH3-C6H4-NO2 -KMnO4/H(+)-> HO2C-C6H4-NO2
HO2C-C6H4-NO2 -Fe/HCl-> HO2C-C6H4-NH2
Ortho aminobenzoic acid can be diazotised diluted in an inert solvant and lead by gentle warming to a strange coupling reaction by concomitant
decarboxylation and denitrogenation forming (C6H4)°° diradical species...
If nothing else is present in the media to react, two of those gather to form a linear tricyclic compound, two benzene ring joined by a cyclobutane
ring (Ar=Ar)
Attachment: C6H4=C6H4.pdf (2kB)
This file has been downloaded 1000 times
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
It has been made from o-nitrotoluene 3 times in the literature, but with NaOH or KOH, and the yield is only 13-15%.
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Quote: Originally posted by S.C. Wack  | | It has been made from o-nitrotoluene 3 times in the literature, but with NaOH or KOH, and the yield is only 13-15%. |
Strange, the oxydation of alkyl part of aromatic ring, especialy with a NO2 protective group must be very high even in pushed conditions...conversion
of toluene to benzoic acid is nearly 100%...
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
I was wrong on the number of references; I forgot about the patent, and the Russian article.
The original 1899 BASF patent from o-nitrotoluene and NaOH:
GB189918319
DE114839
The latest (1972) article that I am aware of is this one.
All other references to this that I am aware of are given there. The local library has closed journal access for some time now, anyone interested in
the references in the article will have to rely on Russian good will and Wiley archive access.
This route may be worthwhile for some despite the low yield.
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
Well I see that espacenet only supports full (very long) urls now. Nice of them to change that. Do the manual number search if you want to see the
patents.
So after two years I finally have full journal access again...Z. angew. 385 (1900) [1 part o-nitrotoluene, 1 part KOH, 1/2 part water, 5 hr reflux] if
anyone cares: http://ifile.it/iywpnc3
[Edited on 5-7-2009 by S.C. Wack]
|
|
|
crazyboy
Hazard to Others
Posts: 436
Registered: 31-1-2008
Member Is Offline
Mood: Marginally insane
|
|
I have been thinking about synthesizing anthranilic acid, what about o-xylene to phthalic acid via potassium permananate then phthalic acid to
phthalic anhydride by heating and then any of the methods mentioned by Sauron to anthranilic acid?
Alternatively "oxidation of naphthalene tetrachloride (prepared from naphthalene, potassium chlorate and hydrochloric acid) with nitric acid" can
supposedly yield phthalic acid but I haven't seen any experimental procedures for that either.
[Edited on 6-7-2009 by crazyboy]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Here is the synthesis from "Experimental organic chemistry - principles and practice"
| Quote: |
Procedure
Dissolve 8.0g of sodium hydroxide in 30ml of distilled water in a 100ml erlenmeyer flask containing a magnetic stirrer bar and cool the solution with
stirring in an ice bath. Add the bromine [2.1ml, 6.5g, 41mmol] (CARE!) in one portion and stir the mixture vigorously until all of the bromine has
reacted [look for the disappearance of the brown colouration] and the mixture has cooled to ca. 0*C. Continue vigorous stirring and add all of the
finely powdered phthalimide [5.9g, 40mmol] to the solution, followed by a solution of a further 5.5g of sodium hydroxide in 20ml of water. Remove the
ice bath, allow the temperature of the mixture to rise spontaneously to ca. 70*C and continue stirring for a further 10 min. Cool the clear solution
in an ice bath with stirring (if the mixture is cloudy, filter under gravity before cooling), and add concentrated hydrochloric acid dropwise with a
pipette until the solution is just neutral when a drop is spotted onto universal indicator paper (ca. 15ml should be necessary). If too much acid is
added, the mixture may be brought back to neutrality by adding further quantities of sodium hydroxide solution, but it is better to avoid this by
careful addition of acid in the first instance. Transfer the mixture to a 500ml beaker (foaming occurs in the next stage) and precipitate the
2-aminobenzoic acid by addition of glacial acetic acid (ca. 5ml). Filter off the precipitate with suction, wash the residue with 10ml of cold water
and dissolve it in the minimum quantity of boiling water containing a little activated charcoal. Filter the hot solution to remove the charcoal and
cool the filtrate with ice. Filter off the pure acid with suction, dry the residue with suction on the filter for 5 min and complete the drying to
constant weight in an oven at 100-120*C.
[Edited on 6-7-2009 by DJF90] |
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
If you happen to have o-xylene, maybe you have some sodium azide as well. In Angew. Chem. 45, 536 (1932) a bunch of amines are made using the Schmidt
reaction: aniline, benzylamine, phenethylamine, etc... and the title cpd., from o-phthalic acid. They say that with 4.15 g. of the acid with 10 ml.
conc. H2SO4 and 40 ml. CHCl3 and a temperature maintained at 45-50C by the slow addition of 4.9 g. NaN3, they obtained 2.7 g. anthranilic acid (yield
80%) and 0.1 g. o-phenylenediamine. No workup details; see volume 3 of Organic Reactions.
[Edited on 12-7-2009 by S.C. Wack]
|
|
|
crazyboy
Hazard to Others
Posts: 436
Registered: 31-1-2008
Member Is Offline
Mood: Marginally insane
|
|
No I don't have sodium azide. I plan on separating o-xylene from meta and para xylenes using the procedure detailed in COPAE:
"When the mixed xylenes are treated with about their own weight of 93 per cent sulfuric acid for 5 hours at 50°, the o-xylene (b.p. 144°) and the
m-xylene (b.p. 138.8°) are converted into water-soluble sulfonic acids, while the p-xylene (b.p. 138.5°) is unaffected. The aqueous phase is
removed, diluted with water to about 52 per cent acidity calculated as sulfuric acid, and then heated in an autoclave at 130° for 4 hours. The
m-xylene sulfonic acid is converted to m-xylene, which is removed. The o-xylene sulfonic acid, which remains in solution, may be converted into
o-xylene by autoclaving
at a higher temperature."
|
|
|
manimal
Hazard to Others
Posts: 180
Registered: 15-1-2008
Member Is Offline
Mood: ain't even mad
|
|
Aniline used to be mfgd. by heating phenol in a sealed tube with ammonia. If that reaction is applied to salicylic acid, would anthranilic acid be the
result? I suppose there may be steric hindrance.
|
|
|
stygian
Hazard to Others
Posts: 242
Registered: 19-9-2004
Member Is Offline
Mood: No Mood
|
|
In the scimad library, in one of the dye-chemistry books, they refer to producing (di)methylaniline by autoclaving aniline with methanol. This +
Hoffman-Martius rearrangement + some oxidation should yield anthranilic.
|
|
|
not_important
International Hazard
Posts: 3873
Registered: 21-7-2006
Member Is Offline
Mood: No Mood
|
|
Those reactions require temperatures in the range of 200 to 250 C, and 50 bar or higer pressures.
Salicylic acid would likely decarboxylate, likely you'd have to use ammonium carbamate ( "ammonium carbonate" ) as a source of ammonia and CO2 to
prevent that. At the temperatures required I suspect the para isomer would predominate.
As for the methylaniline route, you left out protecting the amino group and aromatic ring against oxidation, which will add at least two more steps.
|
|
|
atomicfire
Harmless
Posts: 37
Registered: 7-2-2011
Member Is Offline
Mood: shaprening my molecular scissors
|
|
What is the reaction between phthalic anhydride and urea? Under what conditions does this occur?
ban DHMO
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Some time ago I posted some short report about my failure to perform Hofmann rearrangmenet of phthalimide to anthranilic acid and about running
qualitative analysis on the pthalimide http://www.sciencemadness.org/talk/viewthread.php?tid=19662&... Finally I managed to get a successfull reaction with a decent yield.
A nice book named "Organic Chemistry" by Fieser, Fieser (1956) helped me to rule out what is actually going on in the hofmann rearrangement of
phthalimide. The truth is - you can't do Hofmann rearrangement on phthalimide. Phthalimides, like succinimides, can reversibly attract halogen atom,
and they remain phthalimides. Vogel also mentions the hydrolysis, but there are no instructions about it in the experimental part, they just mix
everything together and let the reaction heat itself.
The actual compound undergoing the rearrangement is a base hydrolysis product - sodium phthalamate or sodium 2-carbamoylbenzoate. If you fail to
hydrolyse the phthalimide properly - you gonna get really poor yields of anthranilic acid, you gonna get a clear amber solution with no precipitate,
which is exactly what happened to me.
You should not add more hypochlorite then stochiometric amount, because on heating the unreacted hypochlorite will convert to a more stable mixture of
chloride and chlorate. The latter is a really strong oxidant that will oxidize and chlorinate the benzene ring, lowering the yields. This is why it's
important to titrate the hypochlorite (e.g. iodine-thiosulfate).
US patent 2653971 has some nice instructions on the reaction conditions. I came to the same reaction sequence as the patent author, while instructions
from the rhodium's page look insufficient. So briefly what I did was 14 g Pthalimide + 10 g NaOH + 80 ml water + cooling ... + 150 ml NaOCl + cooling
... heating... + HCl... + Acetic acid -> collect precipitate.
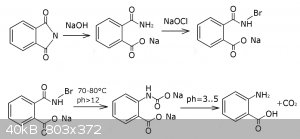

First I dissolved the phthalimide in the 80 ml solution of 10 g NaOH. This gives a pale yellow solution (first flask in the picture). It's important
to make sure the solution temperature is always lower then 20°C, otherwise the phthalimide will be hydrolizing into phthalic acid rapidly (the patent
says the rapid hydrolysis starts at 35°C). This is why you probably want to dissolve it in an ice cold NaOH solution, add phthalimide in portions,
and keep the reaction flask in a fridge. For some reason I could smell a definite odor of ammonia coming off the solution - probably because of some
inorganic ammonium salts like NH4Cl.
After few hours a conversion of phthalimide to sodium phthalamate is complete. Next you should add not more than a stochiometric amount of a cold
hypochlorite solution trying to keep the mixture below 40°C, otherwise the anthranilic acid will start forming, further reacting with hypochlorite,
thus forming chlorinated byproducts. Note that the color of the mixture didn't change. At least I didn't noticed nothing.
Also, patent recommends to ensure the hypochlorite solution was never heated higher then 15°C during it's preparation and storage, but I have no way
to comply the requirement because my hypochlorite solutions all were stored at 20-25°C. So this one probably led to some byproducts and excess
coloration of my final solution.
It's important to keep the alkali always in excess, otherwise the chloramine will react with chloride ions generating a free chlorine, which is
useless for our reaction.
When everything is mixed and exothermic reaction is no longer observed, you can continue to a heating of the mixture. It's possible to use the heat of
chlorination for the subsequent rearrangement, but I'm not that good at this reaction.
Patent tells 60°C is enough, and I have a big research of the Hofmann rearrangement that says only a brief heating is needed for the rearrangement to
complete, although there's no big harm in reaching 75-80°C for a 1-2 minutes to ensure the rearrangement is complete. Thus the reaction mixture turns
pale amber as the patent says (2nd flask in the picture).
I think it's important to pay attention at the odor of the reaction mixture above 40°C. If you have an odor of ammonia - most likely some wrong
reactions are going on. In my last attempt using a freshly regenerated phthlimide I could not notice even a slight odor of ammonia at this step.
Then, after cooling, I carefully adjusted the ph to 7-8 and got almost no precipitate of unreacted something (less than 1 g), because I used at least
stochiometric amount of the hypochlorite. There's a question about what compound it is and should any preciitate be formed at all. I think the
precipitate from rhodium's procedure is unreacted phthalimide, but we ensured it is completely hydrolized.
You should use an excess of phthalimide if you want a reasonably pure product, but I have no idea about how to separate the anthranilic acid and
phthalamic acid (2-carbamoylbenzoic), and nor patent, neither vogel or rhodium says about any recovery instructions.
So the last step is a carefull acidification with acetic acid accompanied by a lot of bubbling of the CO2 coming out of the solution. The solution
turns maroon (third flask in the picture).
If you screwed something and your yield is really poor or not enough acetic acid is added yet, your solution will be clear. But when you have a lot of
free anthranillic acid, it will start precipitating as it does at the fourth flask in the picture.
So, after filtering, washing with water and drying the precipitate I've got 6 g of crude anthranilic acid, corresponding to 45% yield plus copper salt
might give aprox 2 g of more anthranilic acid. Which is almost the same as the yield in the vogel's procedure via hypocbromite and rhodium's procedure
which is an adoptation of vogel's procedure using hypochlorite, though for some reason I could not reproduce the results using hypochlorite. Notice
that vogel's procedure uses freshly prepared hypobromite.

Obtained crystals of anthranilic acid have peru color and seem to have at least few percents of impurities. I'm pretty sure I've got as high as 5-10%
of byproducts, while a 99% compound shall be white colored. Anthranilic acid can be purified by recrystallization from water-detergent mixture. See
"Recrystallization of Organic Compounds From Detergent-Water Systems" Sugihara, Newman, J. Org. Chem. 21, 1445 (1956). DOI: 10.1021/jo01118a032
Or you can just use hot water as Vogel says.
There's a nice qualitative test for anthranilic acid - a solution of Cu(II) (e.g. CuSO4) in dillute acetic acid.

A usual benzoic acid just floats on top as white flakes, phthalic acid doesn't change the solution color (remains blue, not shown on the picture),
anilines give soluble salts too, but anthranilic acid (o-aminobenzoic) gives a green precipitate and m-aminobenzoic acid just gives a blue coloration
(not tested by me). This test also probably will give a green precipitate for chlorinated derivatives of anthranilic acid, like
3-chloro-2-aminobenzoic acid and 5-chloro-2-aminobenzoic acid.
Also, after you filtered off the precipitate of anthranilic acid, you will get a clear maroon solution with a tiny amount of anrthranilic acid in it.
You can precitate it with a Cu(II) salt (CuCl2 or CuSO4). Actually, when I added a CuSO4 solution, a lot of bubles started to come out and I've got
two layers of precipitate (I used a CuSO4 saturated solution): lower gray heavy layer of unknown substance and middle green layer of copper
anthranilate which is much lighter and is really hard to comletely precipitate, so I just filtered it. At first I thought it's a good idea to filter
the solution with a green anthranillate precipitate in it, avoiding the gray bottom layer which is relatively hard to perturb. Latter I found that the
green layer has almost no anthranilic acid. Also most likely I didn't add enough acetic acid and the carbamate didn't hydrolize completely (maybe even
as high as 20% of yield), this is why more CO2 came out on this step and reacted with coper. Thus there are a mixture of some copper salts in the
precipitate.
Next I hydrolized the solid with a NaOH solution with ph >> 12. Solution color changed to turbid green, so I'd say that the very first copper
salt precipitate content was copper bicarbonate, copper acetate and copper anthranilate. After filtering the solid I've got a familiar clear maroon
solution of sodium anthranilate, which I overheated and discarded, so I don't know exactly how much of the product was there, but judging by a solid
left I can say there was up to 4 g (30% yield) of anthranilic acid there.
Another possible reason why I got a poor yield in my first attempts is a dirty phthalimide. Melting point of mine was like 224°C, but after melting
the crystallization point was like 232°C.
The good news are: phthalimide is the most stable of all the phthalic coumpounds in non-alkali high temperature conditions. So you can just add
ammonia solution to anything you've got, except alkaline salts and esters, and after boiling off the water and reaching the temperature above melting
points of all possible phthalic compounds ( 300°C ) you will get a reasonably pure phthalimide. Basically, you can follow the procedure Org. Synth.
1922, 2, 75. DOI: 10.15227/orgsyn.002.0075 http://www.orgsyn.org/demo.aspx?prep=CV1P0457
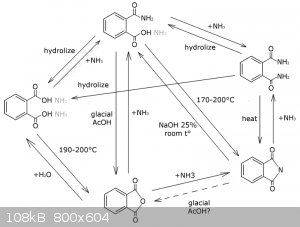
Sources: some internet article and "Concise Encyclopedia Chemistry" By Mary Eagleson (1994), p. 832-833 (you can read at google books)
Aforementioned orgsyn article says you can make phthalimide form ammonium phthalate, but the yield is in question. This can be a nice OTC way to get
the phthalimide derectly from the phthalic acid, either purchased or obtained via hydrolysis of different phthalates.
Amonium carbonate also will work, but I don't know whether urea is a good idea, because the reaction can become violent and the urea can't withdraw
the heat from the mixture unlike ammonium carbonate and water solution of ammonia.
[Edited on 18-4-2015 by byko3y]
[Edited on 18-4-2015 by byko3y]
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
BTW the preparations I've come across that use hypochlorite (obviously one might not want to trust the manufacturer's % statement):
Systematic Organic Chemistry:
40 g of finely powdered phthalimide and 80 g of NaOH are dissolved together in 280 ml of water, the solution being cooled during the operation. The
solution is agitated, and 400 g of a 5% solution of sodium hypochlorite run in. When all is added, the solution is warmed for a few minutes at 80° to
complete the reaction; it is then cooled and neutralised exactly with hydrochloric or sulphuric acid. An excess of strong acetic acid is added to
precipitate the anthranilic acid, which is filtered off and washed with water. Any anthranilic acid remaining in the filtrate is precipitated as
copper anthranilate by the addition of a saturated solution of copper acetate. After standing for some time the precipitate is filtered off and
suspended in a small quantity of warm water, while a current of hydrogen sulfide is passed into the suspension. The copper sulphide formed is filtered
off, and anthranilic acid recovered from the filtrate by concentration on a water bath. It may be recrystallised from hot water.
Total Yield.-85% theoretical (31.5 g). mp 145°.
+
JCE 54, 643 (1977):
Dissolve 16.8 g of NaOH in 50 ml of water in a 500-ml Erlenmeyer flask and cool in an ice-salt bath until the temperature is about 10°C. Add 250 ml
of Chlorox (sodium hypochlorite) and cool to below 5°C. Prepare a solution of 22 g of NaOH in 80 ml of water and cool to below 20'C. Add 24 g of
finely powdered phthalimide in one portion to the cold, alkaline Chlorox solution, and swirl vigorously. Add the second NaOH solution to the reaction
mixture, swirl vigorously, and place on the bench top. Place a thermometer in the flask to monitor temperature. The solid should dissolve as the
temperature rises slowly to about 25°C; the temperature then should rise rapidly to about 50°C. Heat the reaction to 80°C on a steam bath and
maintain that temperature for ~3-5 min. If any undissolved material is present in the reaction mixture, filter at this point. Cool the reaction
mixture in an ice bath, then add concd HCl slowly until the reaction mixture is just slightly basic (wide range pH paper or a pH meter) (about 60 ml
of acid is required). Precipitate the anthranilic acid by slowly adding 25 ml of glacial acetic acid (Caution: reaction mixture tends to foam at this
point). Collect the product on a Buchner funnel and wash with small portions of very cold water until the odor of acetic acid is no longer detectable.
Air dry the solid until the next laboratory period. The yield of anthranilic acid should he 12-17 g; mp 142-144°C (lit. 144-145°C)). If desired; the
product can be decolorized (NORIT) and recrystallized from water...
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Where have you been all this time? Basicly the first procedure is the same as I posted, and they also completely developed the recovery of copper
anthranilate.
So where did you find it? I don't understand or can't find the source.
The second preparation is the same as at rhodium's page and it didn't work for me.
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
It's one of my books that I scanned. Their references are a 1901 lecture involving indigo from naphthalene:
https://books.google.com/books?id=voU3AQAAMAAJ&jtp=139
and the 1890 BASF patent DE55988 which they pretty much translate. The JCE authors adapted Vogel's hypobromite.
|
|
|
Templar
Hazard to Self
Posts: 82
Registered: 17-8-2014
Location: The Sprawl, Titan
Member Is Offline
Mood: No Mood
|
|
Anthranilic acid is good, but if we're going to bother going to quinazolinones, we might as well look at processes where chemicals are less watched.
Phthalic acid is a good starting point for getting either anthranilic acid or isatoic anhydride.
Anthranilic acid still requires the moderately hard to get acetic anhydride, and if a good yield is desired POCl3 or PCl3/PCl5.
I have been investigating synthesis of isatoic anhydride, I posted this here http://www.sciencemadness.org/talk/viewthread.php?tid=33344 in regards to that. However, I didnt realize at the time that the precursor,
phthalimide, had a melting point within 5 degrees of isatoic anhydride, so I wasnt really able to tell what kind of purity I had.
If one could get decent isatoic anhydride, several journal entries and two synthesis reports confirm that ring opening with an aromatic amine followed
by ring closure with an acetylating agent (acetylacetone) would work to produce any range of quinazolinones.
Anthranilic acid is a bit of a strange beast, I didnt realize how annoying it can be dealing with zwitterions until I met this one. The basic form of
anthranilic acid seems to be a brown oil (confirmed by another patent as well) which can solvate in non polar solvents. The amine salts of anthranilic
acid also appear to have a decent solubility in water.
I came across a bird repellent that reportedly claimed 40% methyl anthranilate with 60% veggie oil. This was mixed with 20% HCl soln, resulting in a
suspension of what I believe is the amino HCl salt of anthranilic acid, the oil, and water.
After filtering, this was dissolved in a NaOH solution of pH 11.6 and a milky opaque solution with red/brown oil droplets appeared. I thought washing
with hexane would remove residue veggie oil; turns out it also removed anthranilic acid.
Re acidifcation of the aqueous gave no ppt, but re-combinging with the hexane/oil mix on high stir produced fluffy white crystals that were filtered,
dried and melted at 168C.
I would have taken a water insoluble compound like isatoic anhydride over this anyday, but I must still figure out how to test the purity of it.
Hm..
He who fights with monsters should be careful lest he thereby become a monster. And if thou gaze long into an abyss, the abyss will also gaze into
thee.
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
The isatoic anhydride synthesis you posted seems a lot like the synthesis of anthranilic acid (and you did observe some anthranilic acid produced, you
say). How is it different, in terms of conditions and mechanism?
|
|
|
| Pages:
1
2 |









