Hockeydemon
Hazard to Others

Posts: 218
Registered: 25-2-2013
Member Is Offline
Mood: No Mood
|
|
novel analog of nocaine (theoretical)
I was reading about the nocaine analog of cocaine which is easily synthesized using arecoline and a grignard Now arecoline has no regulation and is found naturally in the betel nut which can be found at most Asian markets.
Unfortunately the grignard employed is needs 1-bromo-4-chlorobenzene which is not OTC and I am unaware of any practical synthesis for it. So I went on
to think about other grignards that could be used. Well p-dibromobenzene is easy to get, but I can't find any drug even remotely similar to nocaine
with a bromine in the 4-phenyl position.
So I went on to Fluorine.. 1-bromo-4-fluorobenzenewas found online in seconds OTC. Using that chemical you'd get methyl
(3S,4S)-4-(4-fluorophenyl)-1-methylpiperidine-3-carboxylate (correct? I'm going to term this flocaine from now on) which is very similar to the paxil metabolite (3S,4R) 4(4-fluorophenyl)3(hydroxymethyl)piperidine. Granted they will be metabolized differently, and I can't really tell you
how differently. There is also a 4-fluorocaine analog which is 30x more potent than cocaine according to the cocaine analog wikipedia and the difference between those two analogs
is equivalent to the difference between nocaine and flocaine. The drug meperidineis identical to flocaine with only exception being the carboxylate group is on the 4 carbon instead of the 3 carbon.
I know when it comes down to it all of these similarities are completely irrelevant considering one change in the metabolic pathway can make a
neurotoxic metabolite. I still find it interesting that I couldn't find anything on the chemical I've purposed.
Here is the mechanism for nocaine found on rhodium

Here is the similar paxil metabolite (3S,4R) 4(4-fluorophenyl)3(hydroxymethyl)piperidine
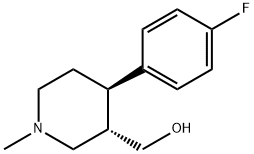
Here is the similar drug meperidine

Here is the drug I'm purposing.
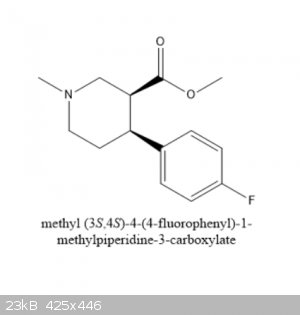
Can anyone shed some light on what I've purposed? Does it exist and I just couldn't find anything on it? Clearly the drug would be an analog of
meperidine which is a schedule II drug in the U.S so making it would be stupid, and trying a completely untested drug would be even more stupid. I
just thought it was interesting. Does anyone know how it would be metabolized in vivo? Since there are drugs that are so similar if I knew what
metabolites would be formed I might be able to figure out more about it.
[Edited on 9-3-2015 by Hockeydemon]
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
You are not aware of practical dihalogenations? Interesting.
Do not confuse chemistry with pharmacology. These are not pharmacologically pethidine analogs, though they may share some chemical similarities. The
similarities are not enough to maintain the pharmacophore. They would still almost certainly be illegal in most locales to produce for consumption,
and arecoline isn't exactly an abundant component of betel nuts, which are banned in many places due to carcinogenicity and even just customs issues
with invasive species.
Pretty much impossible to determine pharmacokinetics without actual data. Even computational programs sold at great expense are very questionable with
the most rudimentary predictions, so don't be fooled into thinking it's easy. Speaking of, lots of these bio isosteres have been made and even tested
in various model organisms. Look into Tropane analogs of cocaine and then piperidine ones. Tons of J. Med. Chem. articles and patents on them. They
tend to have an "RTI" designation.
|
|
|
Hockeydemon
Hazard to Others
Posts: 218
Registered: 25-2-2013
Member Is Offline
Mood: No Mood
|
|
I'm aware of dihalogenations - I could not find any easy way to form 1-bromo-4-chlorobenzene. I searched this forum and found only a couple threads
on the topic. Though if I'm mistaken I am open to your insight. Though I didn't look incredibly hard since I have no real use for it.
Accepting the premise that they are not similar enough to maintain the pharmacophore.. How do you maintain a pharmacophore - not in this instance I
just mean in general. If you move substituants around phenylethylamine based drugs (such as the ones created by Shulgin) there are hundreds of drugs
that are biologically active. So what determines ligand recognition?
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
It's out there.
As for ligand recognition, that depends on the receptor. Some enzymes or receptors are more promiscuous as to substrates, and thus tolerate a wider
range of alterations. The classic way is to test with radiologands, affinity assays, and then develop SAR and QSAR based on efficacy. Newer tools such
STD-NMR make that more definitive. Nowadays, we can work backwards a bit with high fidelity crystallography and NMR data. It isn't really something
amenable to hobbyist in most circumstances.
|
|
|
Hockeydemon
Hazard to Others
Posts: 218
Registered: 25-2-2013
Member Is Offline
Mood: No Mood
|
|
Jesus I'm a fool. Para addition to chlorobenzene with bromine.
I appreciate the clarification on pharmacophore principles. If my drug isn't biologically active & the whole post merely served to demonstrate my
ignorance it can be closed/moved to beginnings.
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
No problem. It is probably active predominantly at dopamine receptors, if at all based on some of the hydrogen data. Fluorine gets interesting.
Sometimes fluorine doesn't substitute well for halogens due to size, and is best to elucidate the type of hydrogen bond donar/acceptor relationship at
an atom, and sometimes it strengthens a bond into a more covalent character through induction, so without checking my drawer of papers tucked in a
filing cabinet drawer on the subject, I am not sure if the data was published, but I am pretty sure it was. Edit- Looks like this is analogous to one
of the WIN compound designations. Behavioral and affinity data is published for that definitively in some models organisms.
[Edited on 9-3-2015 by Chemosynthesis]
|
|
|
Hockeydemon
Hazard to Others
Posts: 218
Registered: 25-2-2013
Member Is Offline
Mood: No Mood
|
|
The only WIN compound I found that seemed similar was WIN 35428 with the only difference being no 2-3 C bridge. It is also similar to troparil though not nearly as similar as WIN 35428.
It does appear that there is a bit of data on various p-C6H8X substituents under Phenyltropanes
I wonder if my drug would metabolize into anything similar to MPTP.
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
There are a few studies generalizing substituent effects on various enzyme affinities relevant to this in the literature, though I would not take them
as definitive by any means.
Looking at p-fluoroderivatives of MPTP does indicate close affinity to MAO-B, comparable to the parent compound, even though this is not an answer to
your question. Comparison with other halogens is more interesting, and there is a lot of primary literature data on these types of compounds,
including pharmacokinetics. Why you are looking through Wikipedia is a bit confusing if you are going to use possessive pronouns with regard to a
compound. Many of these are patented, so it might not be 'yours' in intellectual property, certainly wasn't in discovery, and most probably wouldn't
be in use since these would be viewed as cocaine analogs.
Are you worried about any particular phase of metabolism? A mechanism? Decarboxylation? Proton abstraction? Both? Or is this, as I suspect, a
generalized fear?
By the way, you left out the 4-para substituent when you said the only differences between pethidine and 'fluorocaine' are the ester positions. This
is a huge distinction because 4-para substituents reduce MOR affinity and shift towards serotonin in pethidine (probably also very relevant here on
closer look), and 3' and 2' substituents are generally categorized in addition to the 4'-ester, not as an alternative to it. Still doesn't address
metabolism and toxicology.
[Edited on 10-3-2015 by Chemosynthesis]
|
|
|
Hockeydemon
Hazard to Others
Posts: 218
Registered: 25-2-2013
Member Is Offline
Mood: No Mood
|
|
I know it's not 'my drug' - it's just an easy way to reference it in this context. No fear or worry - just a curiosity. I'd never even consider
consuming a drug that wasn't very well tested. I spend too much money and work too hard on my mind to throw it all away on something stupid like this.
Neuropharmacology genuinely interests me and I easily retain the information presented to me in an exchange such as this.
I would like to know how do you hypothesize the metabolic pathway of something. Say you know to what receptor something binds.. How do you determine
what enzymes your body will use given certain functional groups? Is there an order or priority like there is in chemistry or is it dictated more by
gene expression?
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
Simple question with very complex answer, so I will have to make a long, rambling post. Skip to the numbered section for a brief answer. I think
anyone reasonable would agree with me in essence when I say that you don't really know what metabolic enzymes will function at what proportion in a
human being until you get solid clinical trial data. By this, I mean good patient sampling and duration of observation. This is part of why so many
drugs fail late in production. There are advances in the big data "metabolome" and the "toxidrome" that may eventually make this easier (and probably
will solely out of necessity to reduce drug development costs). Being familiar with metabolic enzymes, I could look at your proposed molecule (scared
me a bit when you say you'd refer to as fluorocaine and then said yours), and give you an educated guess based on known enzymes as to whether it would
produce an MPP+ metabolite. I could actually feel pretty confident in my answer based on homologs and some fancy computational work, but as much as I
would like to hype my own ego, I don't know for a fact.
You can have a good idea of what enzymes do based on animal studies and some cell line data, but there are differences between those and whole people.
Even in human patients, there is variability between/among individuals in terms of things including SNP mutations, CYP expression, food/drug-drug
interactions, age, volume of distribution (body mass index), gender, etc. Careful monitoring is really the only reliable way to ensure you know
metabolites to some degree of confidence. It's true with all enzymes, channels/pumps, various other types of receptors, targets. Judging off-target
effects can be difficult. You can get a somewhat reasonable guess computationally, but these tend to be based off of assumptions. It is easy to train
your model inappropriately, or not test the proper proteins, or not comprehend a metabolite, ad nauseum. It can be easier to look and see what is not
likely, such as a specific transformation, but determining approximate percentages and identities of metabolites a priori is very shaky. Sometimes
this can change with time as enzymes are induced or inhibited.
Even the distribution of a drug in the body can be subtly different from an expectation based on transporter/channel/pump substrate affinity, or just
route of administration (the latter also having some effects on metabolism). New receptors and isoforms are discovered still, and some transient
intermediates may not be detected in the body. No one will want to admit this, but it is also theoretically possible that some methods of metabolite
detection both in vitro and vivo are lacking, as volatile metabolites may not be detected, or a column may not be adequate for separating one out, or
it may just not be at a concentration for an instrument to measure.
Rough generalization for determining what major enzymes are expected to work on a drug usually use pharmacophore type modeling or structure activity
relationships on the metabolic enzymes. Once you have characterized a particular lyase, methyltransferase, transaminase, cytochrome p-oxidase, etc.,
it is much easier to determine how likely a specific molecule is to be a substrate for it... however, given the sheer complexity of the pathways
involved, which can be non-linear, full of feedback loops, diverge and reconverge, and then change with time, it gets extremely difficult.
This generally results in the following type of experimental design: do as much testing up front to determine probable toxicities as early as possible
to avoid wasting time and money. This uses cell lines, lysosomes, and then animals. For the FDA, you generally have to show that 10% or less systemic
exposure of a metabolite occurs to argue for permission to ignore it for further testing and proceed. In this case, the metabolite is often assumed to
be too localized at the site of metabolism (often the liver in pharmaceuticals, though there are inhalational and percutaneous drugs which you would
question this validity to an extent, as well as gut metabolism in some circumstances) to bother with a test, and should produce obvious toxicity where
metabolized if an issue.
If systemic exposure is higher than this, the risks increase and you have to determine new tests per applicable metabolite. Sometimes this is in
animals that produce the homologous upstream metabolic enzyme in comparable concentrations to humans, and sometimes this is in human cells. It can be
in both.
Now as to what determines the enzymes/metabolites themselves?
1) location, location, location. Where does your drug go? What is expressed there?
2) chemistry. Generally, metabolism is taken in phases of biotransformation that act to make a xenobiotic more polar so that is is efficiently
excreted. Oxidation, reduction, conjugation, etc. do follow chemical rules. You can generalize that esters and ethers are cleaved, sometimes alkyl
groups are oxidized to acids, some of the drug may then be conjugated to a more polar moeity for easier elimination. Enzyme expression can change,
which is the next issue. You can sometimes saturate some of these competing enzymes and then things change too.
3) genetics. Not just your genome, but your epigenome or transcriptome too. Classic examples include the "Asian flush" with alcohol, or gerontology
related pharmacology.
This complexity is a big part of why it is easier to repurpose old drugs, develop a "me-too" drug, or develop a new drug for a known target, in
ascending order of difficulty, than to develop something completely novel. It also factors into why post-marketing surveillance is necessary and
carried out.
|
|
|
Dr.Bob
International Hazard
Posts: 2777
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
Just out of curiosity, I looked at this compound, there are at least 8 patents that cover it, with preparations, biological data, and more. Most are
available in Google patents, so not too hard to find free. SKB has many of the patents, so it was likely made during the Paxil years.
BTW, if you are going quote Wikipeadia, do it clearly:
"4'-Fluorococaine is a tropane derivative drug which is a synthetic analogue of cocaine. Unlike related compounds such as the corresponding
4'-fluorophenyltropane derivative CFT and the 2'-hydroxy analogue salicylmethylecgonine, 4'-fluorococaine has only around the same potency as
cocaine as an inhibitor of dopamine reuptake, but conversely it is a much stronger serotonin reuptake inhibitor than cocaine, resulting in a
significantly altered pharmacological profile in animal studies."
The serotonin reuptake activity is not what cooks are looking for... You can get that in prozac... So in the dopamine area, the F and H are
nearly equivalent in activity.
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
Not always the case; MDMA has stronger SERT than DAT affinity.
|
|
|
cmos6667
Hazard to Self
Posts: 50
Registered: 10-4-2015
Member Is Offline
Mood: No Mood
|
|
why not go straight for the real cocaine analog by leaving the benzoyl ester as-is? you'll be 2 carbon short of cocaine and more likely to have the
real deal this way
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by cmos6667  | | why not go straight for the real cocaine analog by leaving the benzoyl ester as-is? you'll be 2 carbon short of cocaine and more likely to have the
real deal this way |
And you're basing that likelihood on what published literature? The tropane bridge is important for dopamine potency, but more amusingly, the benzoyl
ester is not (nor is it even necessarily recommended). Did you even look at the structures discussed? Where exactly do you recommend "leaving" the
benzoyl ester in his molecule? The spot where the Grignard addition takes place? Smooth.
[Edited on 15-4-2015 by Chemosynthesis]
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
Who needs betelnuts for arecoline?
Are we chemists or are we not?
You can get arecoline from nicotinic acid, in three simple steps.
Esterification, quaternization with methyl iodide(methyl tosylate should work too), and then reduction with a borohydride based reductant.
Sorry for necroposting, but I think its much better than starting a new thread.
Attachment: nicotinic acid to arecoline.pdf (491kB)
This file has been downloaded 276 times
Attachment: DARK classics arecoline.pdf (643kB)
This file has been downloaded 330 times
verrückt und wissenschaftlich
|
|
|
Fery
International Hazard
Posts: 1035
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
karlos³ exactly !!!!!
thx for sharing the info about synthesis
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
You're whale cum 
verrückt und wissenschaftlich
|
|
|










