| Pages:
1
..
7
8
9
10
11
..
13 |
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Here is one example US4084995
Attachment: US4084995 Preparation of a cap sensitive particulate explosive composition comprising calcium nitrate.pdf (1.2MB)
This file has been downloaded 971 times
|
|
|
Hennig Brand
International Hazard

Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Thanks for the patent.
I just wanted to add that the DNT produced still had a slight smell of mononitrotoluene. If this was seen as a problem there are several things that
could be done to push the nitration more into the DNT-TNT region and farther out of the MNT-DNT region. The 10% excess nitration mixture specified
could be increased to 20% or more. The temperature could be increased from 80C to 90C for the 2 hours at the end of the nitration. The 2 hour end of
nitration high temperature "nitration finisher" could also be run for more than 2 hours. There are probably other ways to push the nitration farther
as well.
[Edited on 28-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Here is an interesting patent US2435314 attached
Another interesting patent is GB501034 attached
Attachment: US2435314 TNT Nitration method.pdf (462kB)
This file has been downloaded 831 times
Attachment: GB501034 nitration catalysts.pdf (212kB)
This file has been downloaded 700 times
[Edited on 28-11-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I am quite sure that I have successfully produced TNT using sodium nitrate and 98% sulfuric acid. I have a bunch more pictures to share, but I am
having a little trouble logging onto the forum except using the 4G network and my phone. SO3 was used to soak up water in order to produce the 98%
H2SO4 from less concentrated acid. The sodium nitrate was made from sodium bicarbonate and ammonium nitrate; a slight excess of ammonium nitrate was
used and the two reactants were boiled in water until the smell of ammonia was extremely faint. More or less the same setup as was used to produce the
DNT was used, but I ran into a little trouble with the process. I first made >99% H2SO4 and then I could only get ca. 1/4 to 1/3 of the sodium
nitrate to dissolve in it. In a moment of unclear thinking I added an amount of water necessary to bring the acid down 1% to >98% which only seemed
to increases the solubility a little. In retrospect 98% sulfuric acid is probably about right anyway, but adding water was not the way to achieve it.
In twenty minutes or so I came up with "the plan"; the solid sodium nitrate would be dissolved by putting the nitration mixture into the flask and
bringing it up to the reaction starting temperature (80C). The DNT was kept in a small beaker on a small warming tray and kept in the molten state
enough above its solidification point that it would not plug the glass eye dropper/pipette used to add it to the reaction flask. The solid nitrate did
dissolve at the elevated temperature and other than a few minor issues with DNT solidifying in the pipette everything went smoothly. I will explain in
more detail with pictures and quantities used, etc, when I can log on from a full sized computer.
[Edited on 28-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
TNT Production From DNT, Oven Dried NaNO3 and 98% H2SO4
I was using the article Rosco posted earlier, from "The Journal of Industrial and Engineering Chemistry", which had in it the tabulated results of a
series of TNT synthesis experiments. I used experiment No. 16 as a rough guide. The article was based on MNT to TNT, but I still used the same mass
fractions even though I was going from DNT.
Ammonium nitrate was used to nitrate the toluene to DNT, however, there are a lot of indications that sodium nitrate will not decompose the way
ammonium nitrate does at higher temperatures. Ammonium nitrate decomposition products could cause damage to the intermediates or possibly the products
of the reaction and will lower the strength of the nitration mixture at the very least. It was decided that sodium nitrate would be used for the last
stage of nitration.
From DNT to TNT
Nitration Mixture Composition:
82% Sulfuric Acid
16% Nitric Acid
2% Water
(75% Excess Nitric Acid)
For 1g of DNT:
1g / (182.134g/mol) * 1.75 = 0.00961 moles of HNO3 Specified
HNO3 Production:
mass of NaNO3 needed = 0.00961 * (84.9947g/mol) = 0.8168g
mass of H2SO4 needed = 0.00961 * (98.079g/mol) = 0.9425g
produces 0.00961 moles * (63.01g/mol) = 0.6055g HNO3
Reaction Mixture H2SO4:
mass of H2SO4 needed for nitration mixture= 0.6055g/16 * 82 = 3.103g
mass of H2O specified for nitration mixture = 0.6055g/16 * 2 = 0.07569g
Per 1g of DNT use:
0.6055g HNO3
3.103g + 0.9425g = 4.0455g H2SO4 (anhydrous)
0.07569g H2O
Started off with H2SO4 of >95wt%. From previous experience it was known that 130g of NaHSO4 containing pH down would produce about 25g of SO3 and
1.2g of water by the method used. Since about 260-280g of NaHSO4 was used it was assumed that about 50g of SO3 and 2.4g of water would be produced.
10.67g SO3 to tie up assumed 2.4g water in SO3/Oleum produced
39.33g SO3 left from assumed total of 50g
39.33g SO3 / (80g/mol) * (18g/mol) = 8.85g of H2O it can convert to H2SO4
Assume starting with 95% H2SO4 and are aiming for 99%. There is enough SO3 (39.33g) to convert 8.85g of H2O to anhydrous H2SO4. The next time I would
do it right and add enough H2SO4 to get to 98% acid and not add any water after the fact.
99% - 95% = 4% water needs to be removed
H2SO4 needed = 8.85g / 0.04 = 221.23g of 95% H2SO4 (I added 226g by mistake, but the acid still titrated at over 99% after the acid was added to the
SO3)
95% H2SO4 added: 221.23g
SO3 plus H2O it binds with from SO3/Oleum produced: 13.07g
SO3 left from assumed 50g total: 39.33g
Total Produced = 273.63g of 99% H2SO4
Can Nitrate: 0.99 * 273.63g / 4.0455g = 67.0g of DNT
NaNO3 needed = 67.0 * (0.8168 g NaNO3/1g DNT) = 54.7g
Note:
Then I did something foolish and added 2.7g of H2O to bring the water concentration in the nitration mixture from about 1% to about 2%. This is not a
bad thing in a way, as this amount of water can actually be advantageous for the reaction, but a lot more 98% sulfuric acid could have been produced
by adding more 95% H2SO4 instead of water. This would have allowed me to nitrate much more DNT (84.2g instead of only 67.0g).
Procedure:
As stated above the oven dried sodium nitrate was added to the sulfuric acid and then put in the 1L reaction flask. The temperature was brought up to
about 80C to start the reaction and then the pre-warmed and molten DNT was pipetted in to the reaction flask about 1mL at a time over the course of 2
hours. During the addition time the temperature was kept between 80 and 90C. After the addition was complete the temperature was slowly raised. By the
end of the second hour the temperature was up to 130C and was kept in the 130-135C region for another 2 hours. I think I will use a lower maximum
temperature next time (120-125C) since on reviewing the article previously mentioned looks as though this range generally gives the highest yield of
the purest product.
SO3 Production Pictures:
   
Preparing Reactants: (adding the first bit of NaNO3 to the >99% H2SO4 caused it to fume profusely)
 
[Edited on 29-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Reaction Progression Pictures
         
[Edited on 28-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
TNT Synthesis Post Reaction Work-Up
In total there were 2 hours of DNT addition and 4 hours of heating at elevated temperature to complete the reaction. The crude TNT was collected in
two batches. The first batch, which is reported to be of much higher purity generally (COPAE, etc) is what floated and settled on the top of the mixed
acid. The less pure crude TNT was precipitated from the spend nitration mixture by dilution. Since sodium nitrate was used a lot of sodium (bi)sulfate
precipitated as well once the acid cooled. The mixture of crude TNT and sulfate was filtered out and well rinsed with clean water and then put into
boiling water whereby the sodium (bi)sulfate dissolved and the crude TNT became molten and pooled on the bottom. The crude TNT was agitated with
magnetic stirring for 15-20 minutes while molten to remove water soluble impurities. Some of the first crop of crystals (cake) collected from the
surface of the nitration spent acid was recrystallized from methanol, which formed very well defined crystals and brought the melting point up from
the low 70C range to around 80C. The last picture shows the very pale yellow, well formed crystals, of purified TNT.
Yield of washed and dried crude TNT was 44.6g from the first crop and 29.0g from the second crop, for a total yield of crude TNT of 73.6g.
Theoretical Yield = 67g / (182.134g/mol) * (227.13g/mol) = 83.55g
%Yield = 73.6g / 83.55g * 100% = 88.1%
  
Crude product from first crop washed to remove water soluble impurities.
  
Crude product from spent acid processing.
 
First Picture: First crop on left & Second crop on right
Second Picture: Sample of first crop recrystallized from methanol with m.p. around 80C.
 
[Edited on 29-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Here are some more patents of interest attached
These are not directly on topic but related to sensitization schemes involving nitroaromatics including TNT and its lower nitrated precursors and
analogous nitroaromatics used to sensitize NH4NO3. Small amounts of NH4ClO4 and metal salts of copper, manganese, iron, chromium, or lead can also
sensitize NH4NO3 used for OB mixtures. Ca(NO3)2 and nitroparaffins are also sensitizers. Some of these more sensitive systems are sensitive even to a
weakest strength available #1 blasting cap, and are probably sensitive enough for use as reactive target compositions, stump ejection convincer
applications, beaver dam removers, instant hole in the ground installers, budget soft target demolition tasks, ect.
US4746380 and GB497145 and US3184351
Attachment: US4746380 glycine adduct with ammonium nitrate.pdf (572kB)
This file has been downloaded 802 times
Attachment: GB497145 Glycine Detonation Catalyst Nitronaphthalene.pdf (392kB)
This file has been downloaded 843 times
Attachment: US3184351_Trichlorethylene sensitized NH4NO3.pdf (307kB)
This file has been downloaded 726 times
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
2.0g of TNT Initiated; 7.6mm id Al, 0.25g DDNP & 0.35g of LA (Unreinforced Configuration & ca. 6000psi Loading Pressure)
I initiated 2g of the purified TNT shown in the last picture of the last post. I used DDNP in spite of the fact that the only amount left that I had
previously made was sitting on a filter paper on a bureau with dust bunnies in it, etc. It only weighed 0.25g even with all the months of accumulated
dust. I decided to use it anyway and mixed it with 0.25g of lead azide and pressed that on top of the TNT. Another 0.10g of lead azide was pressed on
top as well for a total of 0.35g of lead azide. A tiny bit of basic lead picrate was also used as a flash igniter. There was a detonation with a
fairly large cloud of black smoke, but it wasn't very loud and the damage indicated low order detonation. A dent was made in one side of the witness
plate and a nice scab was blown off the back however. It reminded me exactly of how picric acid acts when not overdriven hard enough. I have a bunch
of sodium picramate stored, so I may make some DDNP soon and perform a decent test.
     
The dent and scab on the right, in the witness plate shots, are from this test.
 
[Edited on 29-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
You would likely get an easier initiation of TNT using an intermediate coupling charge of something more sensitive like styphnic acid, like a half
gram increment substituted for a half gram of the TNT as a coupling charge and booster. The same would apply when picric acid is difficult to initiate
....using an increment of styphnic acid would have about the same effect as using half again as much initiator. The styphnic acid is distinctly easier
to initiate than even picric acid, maybe requiring about 30% less initiator, and has about the same power as TNT.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Sounds like a decent solution. The TNT did seem like it just needed a little more kick to get it into high gear. Even if it took as much as 0.6 or
0.7g of DDNP and 0.05 or 0.1g of lead azide to initiate TNT, it would still make reasonably sized caps. I did some research early this morning
regarding critical diameters of powdered TNT. I re-found a document I had found half a year ago or so; "Critical Parameters For Detonation Propagation
And Initiation of Solid Explosives". It is even more useful now, because of all the data in it regarding TNT. Here are a couple useful graphs which
should help one design around any critical diameter issues. I have attached the whole document as well. The brief write-up before the second graph
said that the researcher used consistent particle size and 1g/cc loading density for all tests for the graph on the right below.
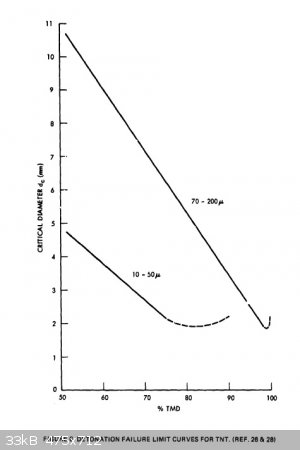 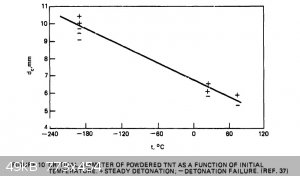
Attachment: Critical Parameters For Detonation Propagation And Initiation of Solid Explosives (2).pdf (4.1MB)
This file has been downloaded 1555 times
[Edited on 29-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Here is a graph with associated equations for TNT & DNT solubility in methanol. Several of the data point values came from the text "Military
Explosives" and a few came from other texts and journal articles. The solubility curves maybe shouldn't be crossing at the bottom, but the graph
should be reasonable accurate at any rate. Attached is also a table taken from "Military Explosives", showing the approximate concentrations of
impurities before and after purification. It is obvious that crude TNT recrystallization from alcohol, or sulfite washes, are not effective for
separating DNTs from 2,4,6 TNT, but they are very effective methods for removing the other isomers of TNT. According to COPAE recrystallization from
concentrated sulfuric acid is the most effective method, of the purification methods commonly used at the time, to remove DNT. However, the best
solution is to run a good strong nitration and convert as much of the DNT to TNT as possible.
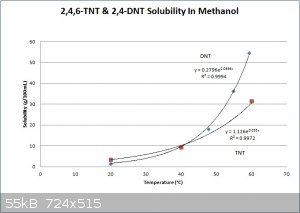 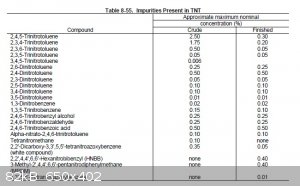
[Edited on 1-12-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
It looks as though the TNT purification method involving sulfuric acid from COPAE is a washing process and not a recrystallization. Apparently
sulfuric acid washes were done as were/are sulfite washes. The crystals of crude TNT are agitated, as a slurry, for an extended period of time whereby
chemical reactions take place with the impurities forming compounds which are much more water soluble and easily removed.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
A couple updates
I didn't post this earlier because I have already posted so much in this thread in the last while.
A few weeks ago I ran another TNT synthesis. A few points of interest:
1. Maximum nitration temperature of 120-125C was used instead of 130-135C and the results were at least visually superior. Even the TNT which was
precipitated from the spent nitration mixture was a nice pale yellow, not orange like the first synthesis where the temperature was taken all the way
up to 135C. I suppose there could be other variables involved as well.
2. For production of DNT from toluene, when I stated earlier that the temperature should be gradually raised from 30C to 80-90C during the course of
nitration mixture addition, that was an oversimplification. Especially during the first and even second nitro group addition care must be taken to
prevent oxidation. Additions must be slow and even and temperature should not be allowed to climb too quickly or nitrogen dioxide will be produced in
unacceptable amounts and oxidation will take place.
3. The best way to push the DNT from toluene nitration farther is to add more nitration mixture (increase from 10% excess to 20% excess perhaps).
Increasing the final temperature from 80C to 90C did not appear to produce any better results at least when held for 2 hours as I did in both cases.
I got a better yield when producing SO3 the last time and made enough 98% H2SO4, from 95% H2SO4, to nitrate about 110g of DNT. Shown below is 76g of
TNT from that batch, recrystallized from methanol, with melting point around 80C. There was much more product, but it was precipitated at a lower
temperature, and some by dilution of the methanol with water, and was not in a dense well formed crystal structure as the sample shown was.

[Edited on 14-1-2015 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Trotsky
Hazard to Others
Posts: 166
Registered: 6-2-2013
Location: US
Member Is Offline
Mood: No Mood
|
|
I have made some MNT using ammonium nitrate and toluene, following basically the same procedure I used for nc, though with important alterations, such
as allowing the temp to remain much higher. I was worried about runaway in doing so, but either a runaway nitration of toluene is difficult or I was
lucky.
I washed and rinsed with bicarb, and added it to a stronger nitration bath and allowed it to sit overnight. I plan on bringing the temp to 90C for
2hr and then cooling. Will this be sufficient to arrive at a reasonable yield of DNT?
My intent is to leave it there until I obtain WFNA or using it to sensitize ANFO enough that tannerite will be able to detonate it.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
If you could post the quantities and reaction conditions used it would be much easier to help you. I will offer the following though, the nitration of
toluene to MNT, or even DNT, does not require the strongest acids and if run properly is a very efficient process resulting in very high yields with
only a slight excess of nitric acid/nitrate. Check the stoichiometry or, even simpler, just look at the quantities I used a page back in this very
thread.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Trotsky
Hazard to Others
Posts: 166
Registered: 6-2-2013
Location: US
Member Is Offline
Mood: No Mood
|
|
I'm away from my notebook but I can get you numbers if you want. I used 10% excess for the first nitration, and 20% for the second.
I wasn't sure if allowing it to sit in the nitration bath overnight before applying heat would hurt anything. This morning, however, before I went to
apply heat I noticed that a mass of beautiful hairlike crystals had formed in it, descending down from a thin layer of the waxy stuff I produced in my
first nitration. Obviously impossible to say for certain, but do you think this is just recrystallized MNT or has DNT formed without applying any
additional heat beyond that generated when the MNT was added to the bath? I delayed the heating phase because I want to get a good photo of these
crystals, they're the biggest I've made of an EM, about 2-2.5 inches in length, but ridiculously thin.
|
|
|
Trotsky
Hazard to Others
Posts: 166
Registered: 6-2-2013
Location: US
Member Is Offline
Mood: No Mood
|
|
From MNT to DNT seems to have failed. The solution turned clear and a small, viscous layer formed on the surface, while small bubbles bubbled up.
This was after 2hrs at 80C. Was this because I used AN and not KNO3? Did the AN decompose?
When the solution was poured over ice chips a very light yellow waxy looking stuff appeared and the solution turned milky and opaque, very unlike the
clear solution I had before.
The almond smell is gone, but is this waxy stuff DNT? Melting pt test time I guess.
Edit: Actually, hennig, the product I had after.letting the MNT sit in nitration bath over night looked exactly.like your picture number two above.
I then took that and heated it to 80C for two hours. Mistake?
[Edited on 19-1-2015 by Trotsky]
|
|
|
TinkerKABOOM
Harmless
Posts: 2
Registered: 19-1-2015
Member Is Offline
Mood: No Mood
|
|
Can you add ammonium nitrate,sodium nitrate or potassium nitrate to the nitric acid to make a more stable yet powerful form of RDX?
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
No. TNT and RDX are completely different, structurally.
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
Ah come on. Throw him a bone. There both cyclic right? Even if RDX is heterocyclic. But to be serious he should really read more before posting.
|
|
|
maleic
Harmless
Posts: 20
Registered: 24-12-2014
Member Is Offline
Mood: No Mood
|
|
The most powerful is probably ONC and HNIW now? Whatever, the preparation of these products are too dangerous.
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I wouldn't say that the preparation of ONC or HNIW is particularly dangerous, however, it is difficult and expensive.
Some of the newer articles about energetic salts (eg. bis-dinitroethylnitramine derivatives) indicate a predicted performance that exceeds HNIW and is
about on par with ONC.
Of course, this may be a group of materials that the software doesn't model well...
|
|
|
Hawkguy
Hazard to Others
Posts: 326
Registered: 10-10-2014
Location: British Columbia (Canada eh!)
Member Is Offline
Mood: Body is Ready
|
|
Alright having problems. I nitrated some toluene for two hours at 20 - 25 degrees C. The result is a bright yellow solid, which melts at 30 degrees,
is explosive, and forms nice crystals. I thought it was para - mononitrotoluene but the melting point is too low. I have doubts about ortho -
mononitrotoluene as well because it sinks... Purification/ ideas on comp?
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
Why does its sinking affect your thinking on what it might be? Both o- and p-nitrotoluene has densities above 1 g/cc and should sink. The low melting
point is probably because you have a mix of isomers, or otherwise impure product.
|
|
|
| Pages:
1
..
7
8
9
10
11
..
13 |









