| Pages:
1
2 |
markx
National Hazard

Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
HMX via Bachmann route
Continuation of the experiments with 70% nitric acid in Bachmann process as discussed previously in the RDX synthesis thread:
http://www.sciencemadness.org/talk/viewthread.php?tid=250&am...
To push things a little further I thought to try for HMX synthesis with the low 70% nitric acid. If RDX is well possible, then HMX should also work
acceptably well. And if it does, then it might just deserve its own thread.
As a basis for selecting the suitable synthesis conditions and methods were the following documents:
http://www.google.com/patents/US2983725
http://www.google.com/patents/US2798870
http://www.google.com/patents/US2678927
http://nopr.niscair.res.in/bitstream/123456789/7060/1/IJCT%2013(4)%20404-410.pdf
http://www.chemikinternational.com/pdf/2012/01_2012/CHEMIK_2...
Especially the US2983725 patent seems to offer a rather convenient method for small scale amateur synthesis of HMX without the separation of
intermediate precursors (DPT). As from the viewpoint of industrial scale production, the method described in the patent is not very suitable, since a
large amount of waste acetic acid is generated. It is hard to utililize and regenerate, hence the large scale manufacture tends to opt for other
routes that generate less waste.
For my small scale experiment it poses no obctructions and I happily opted for the synthesis procedure described in US2983725 with the amount of
acetic anhydride corrected to compensate for the extra water in the 70% nitric acid that I use.
Experimental part
Into the thermostated reactor at 44C were added as heel:
15,7g Glacial acetic acid (GAA)
0,26g Ac2O
0,34g Paraformaline

Magnetic stirring was applied.
The following liquid feeds were prepared:
10,29g Ac2O
4,66g Ammonium nitrate (AN) + 6,17g HNO3 70%
2,02g Hexamine + 3,3g GAA
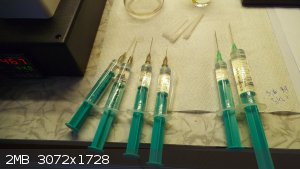
The liquid feeds were added to reactor in 4 portions during 15min at 44C.
1,33g Hexamine in GAA + 1,93g Ac2O at a time. After all the hexamine solution had been added, another portion of 2,56g Ac2O was
added in bulk.
DPT was formed gradually as a white precipitate:
 
The reaction mixture was aged for another 15 min at 44C.
As the second stage (conversion of DPT to HMX) the following liquid feeds were added to the reactor at 44C:
9,6g Ac2O + 5,42g AN in HNO3
The addition was as follows:
Gradual and proportional addition of 6,4g of Ac2O and 5,42g AN in HNO3 in approximately 15minutes, followed by 3,2g of
Ac2O in bulk.
The DPT layer was dissolved quite rapidly and almost completely during the first additions of the liquid feeds:
 
After about 20 minutes from the start of the second liquid feed stage a gradual appearance of another white precipitate was observed (HMX):
 
The reaction mixture was let age for 60 min this time, at the end of which the reactor was filled with a thick slurry of white matter (supposedly
HMX).
7g of water was added to the reactor in bulk and the temperature brought up to 98C for 30min. The digestion stage will purify the product by
hydrolysing some side products.
After the purification, the reactor contents were crashed into 500ml of cold water and the formation of a thick voluminous precipitate was observed:
 
[Edited on 6-8-2014 by markx]
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
The precipitate was filtered, neutralised with sodium bicarbonate solution and washed with coupious amounts of water:
  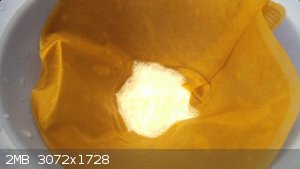  
After being squeezed dry with paper dowels and dryed at room temperature for 24 hours, the final yield was 2,8g of HMX. That translates to 66,5% yield
if my calculations have not failed on me...
Exact science is a figment of imagination.......
|
|
|
Bert
Super Administrator
Posts: 2821
Registered: 12-3-2004
Member Is Offline
Mood: " I think we are all going to die. I think that love is an illusion. We are flawed, my darling".
|
|
Looking forward to watching this thread develop!
As the most difficult reagent for an amateur to acquire is often acetic anhydride, my curiosity caused a quick scan of the procedure & yield.
ROUGHLY 10 grams of anhydride was used per gram of HMX produced... If product was pure HMX.
Rapopart’s Rules for critical commentary:
1. Attempt to re-express your target’s position so clearly, vividly and fairly that your target says: “Thanks, I wish I’d thought of putting it
that way.”
2. List any points of agreement (especially if they are not matters of general or widespread agreement).
3. Mention anything you have learned from your target.
4. Only then are you permitted to say so much as a word of rebuttal or criticism.
Anatol Rapoport was a Russian-born American mathematical psychologist (1911-2007).
|
|
|
Praxichys
International Hazard
Posts: 1063
Registered: 31-7-2013
Location: Detroit, Michigan, USA
Member Is Offline
Mood: Coprecipitated
|
|
Your yield might not be a whole lot of HMX.
HMX is produced by the nitration of hexamine with ammonium nitrate and nitric acid in an acetic acid/acetic anhydride solvent at 44°C. The raw
materials are mixed in a two-step process and the product is purified by recrystallization. This is a modification of the Bachmann Process used to
produce RDX, another explosive. The yield of HMX is about 55-60%, with RDX as an impurity. RDX produced by the Bachmann Process usually contains about
8-12% HMX as an acceptable byproduct.
Do you have a means of analysis? Melting point depression could tell you how much RDX is in your HMX. Density might also be another option.
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Unfortunately I have no reliable means for an in depth analysis at the time to comment on the purity or composition of the product. But constructing a
melting point measuring apparatus could be a good starting point. I will also try to revive my old contacts in the university, perhaps I can gain
access to machinery that allows for a more reliable analysis of the topic.
Exact science is a figment of imagination.......
|
|
|
Dany
Hazard to Others
Posts: 482
Registered: 3-8-2013
Member Is Offline
Mood: No Mood
|
|
For Markx and Praxichys,
there is a fast solution to determine if at least there is one or a mixture of compounds. Do a TLC plate. For this, dissolve the compound in hand in
acetone and spotted on silica gel plate. The plate are developed with acetone-benzene eluant. The HMX and RDX are separated and detected as dark spots
under 254 nm UV light. From the number of spots and the intensity under UV light you can at least estimate how pure is your sample. The procedure
described here can be found in the following document page 3. This procedure is specific for RDX and HMX mixture.
P.S. if you don't have benzene than try toluene.
www.dtic.mil/dtic/tr/fulltext/u2/a155983.pdf
Dany.
[Edited on 5-8-2014 by Dany]
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
You can also find its density? Assuming only RDX and HMX exists in the final product, measuring the density shall tell you everything
|
|
|
Dany
Hazard to Others
Posts: 482
Registered: 3-8-2013
Member Is Offline
Mood: No Mood
|
|
I don't advise the density method. This method is only applicable if one is sure that the sample contain only RDX and HMX. The presence of impurities
will lead to erroneous determination of the composition. Also, this method will not give accurate value if one compound is very minor relative to the
other. The first thing a synthetic chemist do is to check his réaction média or the isolated products with a TLC plate. NMR or other analytical
studies should be performed latter.
Dany.
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Yeah I agree that finding the density might not be a good determination. I wonder what is the smallest cheapest NMR or Spectrophotometer we can
purchase?
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
Have you considered starting from hexamine dinitrate? If it helps with RDX, maybe it can help with HMX.
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
For the sake of scientific curiosity it can be easily tried out, but for practical purposes I personally see little gain in introducing 2 nitro groups
into the setting via a hygroscopic, realatively unstable and poisonous substance (HDN), that adds a whole new synthesis chapter to an already rather
complicated system. Aside from that I do not recall having seen an HMX synthesis based on HDN...might be wrong though.
Exact science is a figment of imagination.......
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
Winkler posits that the Bachmann process actually forms hexamine mononitrate as an intermediate, while the direct nitrolysis forms the dinitrate
intermediate.
In direct nitrolysis, substituting HDN for hexamine is intended to help the yields in the absence of optimum quantity or quality of reagents, like how
you are substituting 70% HNO3. There is less demand for concentration when two moles of -NO3 are present.
Edit: I've included a paper by Winkler when he wrote up his results for differential hydrolysis to measure percent HMX in RDX.
[Edited on 7-8-2014 by roXefeller]
Attachment: differentialhydrolysis.pdf (112kB)
This file has been downloaded 990 times
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Quote: Originally posted by roXefeller  | Winkler posits that the Bachmann process actually forms hexamine mononitrate as an intermediate, while the direct nitrolysis forms the dinitrate
intermediate.
In direct nitrolysis, substituting HDN for hexamine is intended to help the yields in the absence of optimum quantity or quality of reagents, like how
you are substituting 70% HNO3. There is less demand for concentration when two moles of -NO3 are present.
Edit: I've included a paper by Winkler when he wrote up his results for differential hydrolysis to measure percent HMX in RDX.
[Edited on 7-8-2014 by roXefeller] |
Yes, of course you are right about the lesser need for dehydration agent when using the NO3- incorporated into the HDN structure, but what I meant
under "practical purposes" is the fact that this decrease for does not come for free. Ultimately the NO3- has to come from nitric acid, albeit not a
highly concentrated one and you need to separate and dry the HDN (probably under vaccuum or dessicant to separate the water completely) whilst dealing
with its poisonous nature. All that adds up to a lot of extra work which can be avoided with a little extra Ac2O. But that is a matter of choice,
taste, curiosity and scale....in principle I do agree with you on the concept 
BTW...does anyone have an opinion on the feasibility to try the TLC method with filter paper as the column medium? I do not have the original silica
coated plates available, but perhaps a strong porous paper can serve as the separation medium as well.... The retention constants will be different
though, so the direct comparison with official results could be problematic, but at least we should get an idea of the number and extent of components
in the product?
[Edited on 7-8-2014 by markx]
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Well gentlemen....I constructed a makeshift melting point determination apparatus and ran the whole lot of substances that I had in my toolbox through
it. The result were following:
1) ETN, recristallised from ethanol: 62-63C
2) PETN, mixed acid synthesis, recristalised from acetone: 141-142C
3) RDX, Bachmann route, full fraction of product. not recristallised: 197C
4) RDX, Bachmann route, first fraction obtained by cooling of synthesis mixture, not recristallised: 203-205C
5) HMX, Bachmann route, recristallised from acetone: 282-283C
There seems to be a systematical bias of few degrees upwards...not a surprise with an uncalibrated K-thermocouple. But all in all the values add up
and the HMX seems to be quite pure too
Some photographical evidence:
   
BTW, I will have to repeat the HMX synthesis and measure the mp before and after recristallisation. Right now I have only a recristallised sample left
from the first synthesis. I can't comment on how much product was lost by the recristallisation process because I messed it up. I always hot filter
the recristallisation solution to remove any insoluble particles from it before the actual recristallisation occurs via cooling or dilution. But with
HMX this proved to be near impossible....the acetone solution immediately cloggéd the filter paper and not a single drop came through. I tried
several types of papers, but all I accomplished was a considerable loss of solution that was soaked into the filter paper and became unredeemable.
Hence I crashed the remaining solution directly into water and saved what I could. I did not even bother to weigh the result...it would not have told
me anything reasonable in terms of process yields.
I will try the TLC plate method too, but first I must perform another synthesis to get a representing sample of product. I will recristallise most of
it and leave a small sample for analysis from the raw alpha HMX directly from synthesis. That way we can get a nice comparable result on the effect of
recristallisation and the yields and of course the number and rough estimation of content of products formed.
[Edited on 11-8-2014 by markx]
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
I did try the TLC method using filter paper as the column media and 50/50 by volume mixture of acetone/toluene as the eluent...it's a no go. The
filter paper is absorbing the eluent at a relativistic speed and everything is rushed up by the solvent front...no reasonable separation is achieved.
I rigged up a UV-c germicidal tube to light up any information on the TLC plate, but I could not make out anything of useful purpose. I guess I'll
have to order some proper TLC plates from ebay or try to make some DIY ones from microscope slides.
Is there any other reasonably realised methods for developing a TLC plate with nitramines besides the UV radiation? I did find some methods on the
web, but none of them seem to be very suitable for the TLC plate.
Exact science is a figment of imagination.......
|
|
|
Praxichys
International Hazard
Posts: 1063
Registered: 31-7-2013
Location: Detroit, Michigan, USA
Member Is Offline
Mood: Coprecipitated
|
|
This is pure speculation, but perhaps if the plates were slowly heated, varying decomposition temperatures of some compounds might be evident?
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
| Quote: Originally posted by Praxichys | | This is pure speculation, but perhaps if the plates were slowly heated, varying decomposition temperatures of some compounds might be evident?
|
I think if a paper substrate is used, the decomposition of cellulose will appear before any of the nitramines show signs of alteration. Anyways, the
fisrt step should be the aquisition of proper TLC plates. Constructing the needed tools for any job will always draw attention away from the objective
and make the whole process very tedious. Therefore I have begun to accept the philosophy of buying the tools instead of making them (at least the
simple ones)
As an example of the opposite I did construct a drop tester:
   
A simple design with a quick release dropping a 2kg hammer from the preset height onto a sample packed in Al foil. Works like a charm.
The results:
ETN: 30cm
PETN: 40cm
RDX Bachmann route mp 197C: 55cm
RDX Bachmann route mp 203-205C : 65cm
HMX Bachmann route mp 260C: 78cm (a single partial detonation at maximum height of coulmn, I could not replicate the result)
HMX Bachmann route mp 282C*: - no go at maximum height of 78cm with 2kg weight
* I must note a correction to the melting point of recristallised HMX sample from first synthesis. The corrected mp is 270C and there is clearly
another component(s) in the product (supposedly RDX). The measuring technique makes all the difference in the final result. At first go I supplied the
sample of HMX onto a relatively cool (150C) measuring apparatus and raised the temperature slowly until it reached the 282C mark and the sample melted
abrubtly. But that was all wrong, because what I actually did, was to evaporate and decompose all the impurities in the sample as I raised the
temperature slowly and was left with the highest melting component in the end...HMX. A simple distillation if you will. Since the sample was small I
did not notice the other components leaving it. After realising my error, I heated up the apparatus to the expected mp and then placed the sample on
to the machine. This revealed the corrected values for HMX samples.
Not recristallised alpha form of HMX, directly from syntesis: 260C
The sample softened at 255C and melted at 260C after which the impurities quickly boiled off and the sample resolidified until 282,7C was reached.
Recristallised sample of beta HMX from first synth: 270C
Sample softened at 260C and partly melted at 270C (crystals became transparent and a liquid phase was formed). Again the impurities boiled off quickly
and sample resolidified until 282,7C was reached.
I also remeasured the mp of the rest of substances mentioned in previous posts ( RDX, PETN, ETN)...their mp-s remained the same.
Exact science is a figment of imagination.......
|
|
|
Jimbo Jones
Hazard to Others
Posts: 102
Registered: 15-10-2009
Member Is Offline
Mood: No Mood
|
|
Excellent work. Thanks for sharing.
|
|
|
Vpatent357
Harmless
Posts: 22
Registered: 10-7-2014
Member Is Offline
Mood: No Mood
|
|
i like it, can you make a hammer test drop with SA.DS for show, pm me 
Thanks
Small blasting caps, fingers safe!
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
It will be a short fall and a loud bang...to gain any systematic knowledge about the matter of SADS a series of experiments is in order. Different
synthesis conditions vs impact- friction- and ED sensitivity. Perhaps even some mixtures of SADS with energetic or OB balancing additives, even though
the mixture part fills me with doubt and fear. Perhaps incorporating the additives as a solution would make reasonable sense. Anyhow, I have it
planned for the future, but I am in the midst of constructing the friction sensitivity measuring rig right now. Anybody have any clever ideas for
constructing an ED sensitivity measuring machine?
A set of HV capacitors with varying values, coupled to a low power switch mode dc HV generator would perhaps be the simplest option. The combination
of capacitance and varied spark gap distance can be recalculated into energy, assuming the dielectric properties of air are constant over different
experiments. In reality it can be subject to change (relative humidity). The reliable determination of the energy of the discharge seems to be a tough
part, but without that the results are pretty much worthless.
Exact science is a figment of imagination.......
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Good job. Building a drop hammer device has been on my to-do list for a while now.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
| Quote: Originally posted by markx | | In reality it can be subject to change (relative humidity). The reliable determination of the energy of the discharge seems to be a tough part, but
without that the results are pretty much worthless. |
Can you purge and fill a bell jar-enclosed tester with argon weld gas?
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
| Quote: Originally posted by roXefeller | | Quote: Originally posted by markx | | In reality it can be subject to change (relative humidity). The reliable determination of the energy of the discharge seems to be a tough part, but
without that the results are pretty much worthless. |
Can you purge and fill a bell jar-enclosed tester with argon weld gas? |
Can be done....actually only the electrodes and the sample need to be enclosed in a purge chamber. The rest of the tester can be situated in normal
atmosphere. Although the chamber has to be able to withstand quite a shock.
Freeze frame of a 2-3mg PETN sample going off in the drop tester:

As can be seen from the size of the fireball, the term "energetic material" is well justified.
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
A few frames of the melting point measuring apparatus....I built a more civilised setup with a USB camera looking into the sample well:
    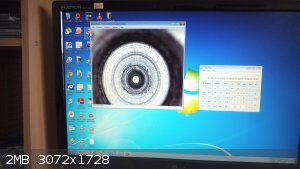
I must admit the heat transfer block from a welder rectifier diodes heat sink is not the best of choices, but it was the only one I had laying around.
I probably will exchange it for a piece of rectangular aluminium stock if I get the chance, the heat loss at higher temperatures is just too big (it
is a heat sink after all). The setup is powered by 220V 100W mould heater. A very compact and powerful heating element that is compatible with my
trusted PID controller.
Exact science is a figment of imagination.......
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
You have built some very neat things. I really like the drop hammer/impact sensitivity tester. For the melting point apparatus I would suggest a
block, or cylinder would even be better, of aluminum with no fins and maybe even some added insulation would be a good idea. What works best is a
large thermal mass. A large thermal mass resists sudden changes in temperature (the thermal flywheel effect), which is what works best when trying to
get an accurate value for melting point. Fins are designed to allow the heat sink to transfer heat/energy to the surroundings as quickly as possible.
I see that you did already say that it wasn't the best choice of heat sink, but I thought I would comment anyway.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
| Pages:
1
2 |









