| Pages:
1
2
3
4 |
DraconicAcid
International Hazard

Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
When you try the iron with en, perhaps try it under anhydrous conditions? That would prevent formation of slimy hydroxides, and en should form a
complex with Fe(II) (based on the fact that Lang's handbook gives a formation constant for it).
The chromium is probably only giving a slimy precipitate because it's non-labile. The complex [Cr(en)3]3+ should be very
stable, just slow to form.
[Edited on 29-10-2013 by DraconicAcid]
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Quote: Originally posted by woelen  | The reason why I prefer to use NH4ClO4 in many cases is that I do not want to use up my HClO4 quickly. NH4ClO4 is much easier for me to come by than
HClO4. If I were in a lab with supply of chemical without restrictions, then I would use the metal oxide or carbonate in all cases and would dissolve
that in aqueous HClO4.
With copper, however, I used HClO4 in which I dissolved CuO. This was because it was written that the copper complex is very soluble and less easily
crystallized and in such cases it is best not to have foreign ions in solution as well.
My next one will be the cobalt(III) complex and maybe the silver(I) complex, although I am somewhat reluctant to make the latter. Silver(I) with
ammonia is a dangerous combination on standing for a longer time and I can imagine that silver(I) with (en) also is dangerous, regardless of the
anion. So, if I try the silver complex, I will try it on a very small scale.
I already tried chromium(III), iron(II), iron(III) and vanadium(IV) and all of these form a slimy/flocculent precipitate with (en), which does not
lead to formation of clear solutions and easily crystallizable compounds.
[Edited on 29-10-13 by woelen] |
I use HClO4 because I have quite a lot of it at hand (70%).
But I don't have NH4ClO4.
Everybody has its own troubles (cross to bare)  . .
By metathesis NH4ClO4 is as potent as HClO4...:
1°)It can turn many metal hydroxyde or oxyde into the desired metal perchlorate or amino metal perchlorate (what is relatily unstable and in open
system free amonia by exchange towards water ligands).
2°)It can turn EDA into EDA diperchlorate and free NH3 as a gas.
There are many complexing metal specific of amines, even calcium (what I was unaware of) or mercury that has a special affinity for it.
Silver is particularly uncompatible with ammonia because of formation of fulminating silver Ag-NH2, Ag2NH and Ag3N (nightmare of the mirror makers);
but other amines will be much less of a trouble... with exception of hydrazine what is immediately decomposed into metallic silver and nitrogen.
It is always a good idea to test in tiny quantity because the potency of the catalytic effect of the metallic core (of not wel documented complexes or
unmade to date complexes) is not clearly known until done and tested towards time decay, temperature sensitivity, shock sensitivity and light
sensitivity.
[Edited on 29-10-2013 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Chemstudent
Harmless
Posts: 44
Registered: 3-3-2012
Location: Sunny Florida
Member Is Offline
Mood: Contemplative
|
|
I've been working on Nickel complexes in my Inorganic lab this semester. It really has been a fun experience. Working with Ni(II) complexes has really
helped me learn the spectrochemical series and colour theory. Although some of these complexes have been super hard to produce (namely the water
sensitive varieties)
I have one this week that I'll be tackling for a second time - [Ni(NO2)2(en)2] which is a red linkage isomer. I begin with (K4[Ni(NO2)6].H20) <--
Hexanitronickelate (II) which is an orange-clay compound. It is very hygroscopic so all the glassware & reagents must be as moisture-free as
possible. Any moisture and you end up getting a purple aqua/nitro complex.
Last thing though, I am still unsure what commercial value or medical/scientific use these linkage isomers have. Is there any use for these compounds?
|
|
|
woelen
Super Administrator
Posts: 8027
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Your complexes, are they nitrito complexes or nitro complexes? If you can give more details on how to prepare them (and if I have the reagents and
needed apparatus), then I certainly would like to try them myself.
---------------------------------------------------------
In the meantime, I now tried the cobalt(III) (en) complex. This is not hard to make, I took a fairly concentrated solution of cobaltous nitrate, but
if you don't have that, you can use cobaltous carbonate and nitric acid to make such a solution.
To this solution, I added quite some excess amount of (en) and swirled. This leads to formation of a reddish/brown solution. In contact with air, this
solution becomes MUCH darker. You can see the dark color near the surface and on the glass.
Next, I added 10% H2O2 while swirling and continued adding this, until there is weak effervescence. Initially, the H2O2 immediately reacts with the
cobalt(II) complex and oxidizes this to cobalt(III). But at a certain point, the H2O2 is not used up anymore and then it slowly decomposes.
I then boiled the liquid for a while to be sure that all excess H2O2 is destroyed.
In a separate test tube I dissolved a slight excess amount of NH4ClO4 in a small amount of water (I needed to heat the water to near boiling). This
solution of NH4ClO4 is added to the cobalt(III)/(en) solution and then the liquid is boiled again for some time, just to be sure that no solid
material is present in the liquid.
On cooling down, a lot of crystalline material is formed. This is dried as described above in the thread. Pictures and a more extensive write-up will
follow one of these days.
--------------------------------------------------------------------
I also tried the silver(I) complex, but this failed.
I prepared a solution of AgNO3 in water and added drops of (en) to this solution. Initially, this leads to formation of a yellow/brown precipitate but
if enough (en) is added, then the precipitate redissolves again and a clear solution is obtained.
To this solution I added a solution of NH4ClO4. When this is done, then the liquid becomes turbid again, it becomes dirty white. On heating, the
liquid becomes more turbid and color of the precipitate turns to light brown and part of the small particles coalesce to somewhat larger particles. No
crystals are formed, a firtly looking water-insoluble precipitate is formed. I think it is a mix of Ag2O, AgOH, some Ag(en) complex and maybe ammonia
complexes. Not something which I wanted to keep around for a long time (knowing the danger of ammonical silver solutions), so I destroyed all of it by
adding excess dilute HNO3, which dissolves all of it to a colorless solution.
[Edited on 3-11-13 by woelen]
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by woelen | | No crystals are formed, a firtly looking water-insoluble precipitate is formed. I think it is a mix of Ag2O, AgOH, some Ag(en) complex and maybe
ammonia complexes. Not something which I wanted to keep around for a long time (knowing the danger of ammonical silver solutions), so I destroyed all
of it by adding excess dilute HNO3, which dissolves all of it to a colorless solution. |
I wouldn't expect the en complex to be particularly stable, since silver(I) pretty much always forms linear complexes, and ethylenediamine isn't long
enough to chelate in a linear fashion.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
woelen
Super Administrator
Posts: 8027
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Ah, that is interesting info you give about the linearly shaped complexes. This also explains why silver(I) usually has only two ligands coordinated
to it (or one bidentate ligand).
I dried the cobalt complex. I think that the cobalt complex also is less stable than other complexes. I obtain a mustard-colored solid, while the
liquid from which it is obtained is much more red (nearly as red wine, albeit with a brownish tinge). The yield is very low, when compared to the
yield of the other complexes.
The solid material is very easy to ignite, it is much more sensitive to flame than any of the other complexes I made. It is not the most energetic
though. The copper complex is more energetic.
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by woelen | I dried the cobalt complex. I think that the cobalt complex also is less stable than other complexes. I obtain a mustard-colored solid, while the
liquid from which it is obtained is much more red (nearly as red wine, albeit with a brownish tinge). The yield is very low, when compared to the
yield of the other complexes.
The solid material is very easy to ignite, it is much more sensitive to flame than any of the other complexes I made. It is not the most energetic
though. The copper complex is more energetic. |
I was about to post that this information is very surprising, as it's an extremely stable cation... but then I remembered you had perchlorate
counterions. When I was a grad student, I assisted with a course that made a set of cobalt(III) en complexes every year, and the tris(en) complex was
an impurity in most of the products.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
woelen
Super Administrator
Posts: 8027
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Could you provide information about the other Co-(en) complexes you prepared in that course and do you have procedures for that? I might be able to
modify these procedures, such that I can make similar complexes with the reagents which I have.
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Alas! this was more than fifteen years ago, so I don't remember the exact procedures. I didn't keep the lab manuals after graduating.
Basically, we made [Co(en)2(NO2)2]Cl by reacting CoCl2, en, and sodium nitrate in a solution through which
air was pulled through overnight (using a vacuum aspirator). Once this was isolated, we added hydrochloric acid to get the green
trans-[CoCl2(en)2]Cl salt (this always looked like crap, because it was contaminated with the tris(en) complex, among other
things). Heating the green complex in water with a steam bath converted it to the purple cis complex. We could also dissolve the green complex in
water and add a concentrated solution of potassium nitrate, which formed the much-less-soluble trans-[CoCl2(en)2]NO3,
which were very nice green xtals (since the tris(en) complex and all the other crap that was in the original green stuff did not form a less-soluble
nitrate).
In basic solution, these would hydrolyze to a pinkish [Co(H2O)2(en)2]3+ ion.
I heard that they replaced the "overnight aspiration" prep with a hydrogen peroxide one, which took much less time. I seem to recall that activated
charcoal was added as a catalyst.
At one point I tried replacing the the chlorides with oxalate (got a red material which might have been the aquo complex) or iodide (Co(III) plus a
reducing agent- you can guess how well that worked).
Some references:
http://www.uiowa.edu/~c004153a/Co%28en%293-2004.pdf
http://chemlab.truman.edu/CHEM131Labs/CoordChem.asp
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
An attempt at tris(ethylene diamine) nickel(II) hydroxide
Yesterday, I made an ethylene diamine nickel complex in the following manner. Half a spatula full of wet nickel(II)hydroxide was put in a test tube
and about 2cm of demi water were added. To this approximately 0.7ml of ethylene diamine were added.
The hydroxide attached to the glass of the tube immediately received a pink-purplish layer on the outside. On the inside it remained green, and in
between was a dark blue region. The hydroxide in the water turned a bit whitish. After shaking and standing for a minute, everything had become
pink-purple, just as the tris(ethylene diamine) complexes produced by Woelen at the beginning of this thread. It surprised me that the liquid on top
of the pink-purple precipitate was also pink-purple. Apparently the newly formed substance is somewhat soluble in water.

The picture was taken under incandescent light, and white balance corrected to represent the colour correctly. Under the light of mercury vapour
lamps, the colour of the pictures became far too blue.
Note that the nickel(II)hydroxide was not entirely pure. It was created by adding an NaOH solution to a solution of black nickel oxide in hydrochloric
acid solution. The hydroxide was used without further purification.
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Nice nickel.
I've seen a sample of [Ni(en)3]Cl2, which was made by accident. The students were supposed to be making
NEt4NiCl4 by dissolving nickel(II) chloride in ethanol, adding triethylformate, and then adding tetraethylammonium chloride.
One student accidentally added ethylenediamine instead of triethylformate (they both said "ethyl" in the name somewhere, you can't blame her for not
reading the entire label....honest!), and got an instant precipitate of a very similar purple material.
I'm not surprised that it's water soluble- many hydroxides of complex ions are perfectly water-soluble, in stark contrast to the simple hydroxides.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
woelen
Super Administrator
Posts: 8027
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Interesting to see that you can get a hydroxide complex as well. This can be a good starting point for many other complexes. It would be interesting
to see how it behaves if dilute sulphuric acid is added very carefully, such that the hydroxide is just neutralized. Does the hydroxide react first,
or is the (en) protonated, resulting in destruction of the complex?
I have done a similar small experiment, by adding some CuO to a solution of (en) in water. If this is done, then the CuO partially dissolves, giving a
blue/purple solution.
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by woelen | | I already tried chromium(III), iron(II), iron(III) and vanadium(IV) and all of these form a slimy/flocculent precipitate with (en), which does not
lead to formation of clear solutions and easily crystallizable compounds. |
I was just looking up a prep for Cr(en)3 complexes- if you add a bit of granular zinc, it can catalyze the replacement of the ligands by reducing the
Cr to the labile Cr(II) state. Try that.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Chemstudent
Harmless
Posts: 44
Registered: 3-3-2012
Location: Sunny Florida
Member Is Offline
Mood: Contemplative
|
|
| Quote: Originally posted by woelen | Your complexes, are they nitrito complexes or nitro complexes? If you can give more details on how to prepare them (and if I have the reagents and
needed apparatus), then I certainly would like to try them myself.
[Edited on 3-11-13 by woelen] |
Sorry for not having returned an answer sooner! I have since been able to synthesize a small quantity of the troublesome Nickel(II) complex which I
had mentioned earlier. And no, it wasn't a nitrito complex. Below I will include any relevant information from my lab manual:
"The intent of this experiment is to extend the student's ability to synthesize, purify, and characterize coordination compounds. In addition, the
ambidentate nature of the NO2 ligand is investigated...."
Three linkage isomers of Nickel(II) were synthesized from the precursor: hexanitronickelate(II)monohydrate,
K4[Ni(NO2)6].H2O
The precursor was prepared in ~40 minutes time, however was placed in a desiccator over an extended period for drying. See attached word document for
detailed procedure. Il a été très facile!
Now...
Two of the linkage isomers found in the experiment were easily synthesized in good yield. One however took me 3 attempts to produce; even then the
yields were quite poor. I have not included the procedure for the other two compounds (pm me if you want the others). Included is the procedure for
producing the compound I found very troublesome.
The complex is: [Ni(NO2)2(en)2] it was found to be ruby red in colour.
The procedure transcribed from my lab manual is attached in a second word document. For this procedure I had not offered additional input, rather I
merely transcribed the directions. It is however important to note the reaction conditions should be moisture free as possible (wet glassware/reagents
was a big issue).
Attachment: Precursor - Hexanitronickelate(II) monohydrate.docx (32kB)
This file has been downloaded 659 times
Attachment: red complex - [Ni(NO2)2(en)2].docx (96kB)
This file has been downloaded 625 times
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Looks cool, Chemstudent. For the precursor, however, it says to make it in de-ionized water, but it also calls for 100 mL methanol (but you only
wash with 15 mL methanol). Is there an error there?
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Chemstudent
Harmless
Posts: 44
Registered: 3-3-2012
Location: Sunny Florida
Member Is Offline
Mood: Contemplative
|
|
| Quote: Originally posted by DraconicAcid | | Looks cool, Chemstudent. For the precursor, however, it says to make it in de-ionized water, but it also calls for 100 mL methanol (but you only
wash with 15 mL methanol). Is there an error there? |
H2O in the production of the precursor is used to help catalyze the reaction. The final product is a mono-hydrate complex, however it will
have more H2O in its coordination sphere which is why the final product must be dried in a desiccator. I was told by my laboratory
instructor that the compound is hygroscopic, thus it must remain in a desiccator for extended storage. Further, it must be in the mono-hydrate state
in order to successfully form the Ni(NO2)2(en)2 complex. As stated, the second synthesis is very sensitive to
moisture, and a wee drop can ruin the yield. H2O in that capacity would act as a catalyst to push out ALL the NO2, and the en
would fully react with the Nickel to form a purple Ni(en)4 complex. What is desired is to only displace 1/2 of the NO2 ligands, and replace
them with the (en) ligand. Controlling not only moisture, but the rate of ethylenediamine addition was equally important. This synthesis really
challenged my understanding of kinetic/thermodynamically favored products.
Lastly, I always state DI water because I never want there to be any confusion as to if other sources of H2O are acceptable. I never use
anything other than DI in lab procedures.
Oh, and the 100mL Methanol was just me saying have it on hand in a wash bottle in case you want to clean the product above and beyond what is
necessary. I ended up using more to wash my product, but only because I am one who likes to go overboard. I wanted to clean off as much impurity as
possible. It only hurt my final yield slightly and that I was not concerned with.
[Edited on 22-11-2013 by Chemstudent]
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
Ethylene diamine with MoO[sub]3[/sub]
I tried to create a molybdenum complex with (en). As anticipated, this was not a success.
Half a spatula of MoO3 (ceramic grade, medium grey powder) was put into a test tube with some 3 cm of demiwater. This gave a whitish suspension on
shaking. On heating the oxide did not dissolve, neither after addition of a few drops of concentrated H2SO4. To the still hot solution, 1 ml of (en)
was added. Immediately the suspension became clear, leaving only the larger particles of MoO3 on the bottom of the test tube. Some of these were
bluish, indicating that the starting material used did not have all molybdenum in the +6 oxidation state.
After addition of another ml of (en) and shaking the mixture near its boiling point for a few minutes, all of the oxide had dissolved into a
completely clear solution, almost colourless, with only a slight hint of yellow.
I do not think that (en) did any complexation to the oxide. Instead, a (poly)molybdate has been formed, with (en) acting as the counter-cation.
MoO3 (s) + H2O + (en) (aq) --> MoO4(2-) (aq) + H3NCH2CH2NH3(2+) (aq)
MoO4(2-) may in turn have complexated with more MoO3 to form a polymolybdate anion, although I think that there was too much (en) in solution for this
to happen.
|
|
|
woelen
Super Administrator
Posts: 8027
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
In this case, (en) simply acted as a base, needed to dissolve the MoO3. Nothing really interesting. Apparently (en) is a strong enough base to
dissolve MoO3.
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by Chemstudent |
H2O in the production of the precursor is used to help catalyze the reaction. The final product is a mono-hydrate complex, however it will
have more H2O in its coordination sphere which is why the final product must be dried in a desiccator. I was told by my laboratory
instructor that the compound is hygroscopic, thus it must remain in a desiccator for extended storage. Further, it must be in the mono-hydrate state
in order to successfully form the Ni(NO2)2(en)2 complex. As stated, the second synthesis is very sensitive to
moisture, and a wee drop can ruin the yield. |
I tried this reaction today- that was interesting. Adding the nitrite gave a deep green solution, and slowly gave an orange-red precipitate. The
filtrate was olive-green, until I washed the precipitate with alcohol and more compound precipitated from solution.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
| Quote: Originally posted by woelen | Thanks [nezza] for this video and picture. Your sample looks very much like mine, so that is a good sign. My sample looks slightly more blue, but the
picture was made in daylight. Under TL-light, the material has exactly the same color as shown by your picture.
(...)
[Edited on 19-9-13 by woelen] |
Funny that these compounds look different under filament/discharge lamp light. Here's the nickel(en)hydroxide, that I crystallised from its solution
under filament lamp light. Though not visible in thepicture, the crystals are glistening a bit in the lamp light.
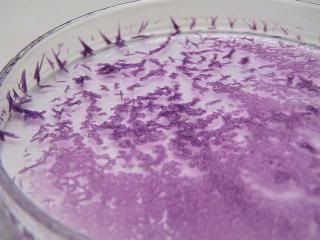
|
|
|
Chemstudent
Harmless
Posts: 44
Registered: 3-3-2012
Location: Sunny Florida
Member Is Offline
Mood: Contemplative
|
|
| Quote: Originally posted by DraconicAcid |
I tried this reaction today- that was interesting. Adding the nitrite gave a deep green solution, and slowly gave an orange-red precipitate. The
filtrate was olive-green, until I washed the precipitate with alcohol and more compound precipitated from solution. |
That's great! So I assume you obtained the Orange-Red Hexanitronickelate complex? It looks like the clay found on a baseball field to be honest. Now
leave it in a desiccator to dry and then you can go from there to the red complex. I will be very impressed when you do, but the final product is
worth it. You get crimson red crystals. They are nice to look at.
[Edited on 3-12-2013 by Chemstudent]
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by Chemstudent |
That's great! So I assume you obtained the Orange-Red Hexanitronickelate complex? It looks like the clay found on a baseball field to be honest. Now
leave it in a desiccator to dry and then you can go from there to the red complex. I will be very impressed when you do, but the final product is
worth it. You get crimson red crystals. They are nice to look at. |
I don't actually have a desiccator, so I tried it briefly in the oven at low temp. I'll let you know when I try the final product. Couldn't compare
it to the stuff on a baseball field, though- I'm not a baseball kind of guy.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Chemstudent
Harmless
Posts: 44
Registered: 3-3-2012
Location: Sunny Florida
Member Is Offline
Mood: Contemplative
|
|
| Quote: Originally posted by DraconicAcid | | Quote: Originally posted by Chemstudent |
That's great! So I assume you obtained the Orange-Red Hexanitronickelate complex? It looks like the clay found on a baseball field to be honest. Now
leave it in a desiccator to dry and then you can go from there to the red complex. I will be very impressed when you do, but the final product is
worth it. You get crimson red crystals. They are nice to look at. |
I don't actually have a desiccator, so I tried it briefly in the oven at low temp. I'll let you know when I try the final product. Couldn't compare
it to the stuff on a baseball field, though- I'm not a baseball kind of guy. |
Well, there may be the issue of pulling atmospheric moisture when cooling down the complex to room temp as it is somewhat hygroscopic. You would do
well to quickly store the dry complex in a container immediately after pulling it from the oven so as to minimize adding moisture. Still shouldn't be
an issue. And by low temp, I hope you mean no greater than 60C
[Edited on 3-12-2013 by Chemstudent]
|
|
|
DraconicAcid
International Hazard
Posts: 4355
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
It was about 50, and I sealed it up immediately after. It still looks nice.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
Bisethylenediamminenickel(II) bistri-iodoplumbate(II)
===A description with follow when I edit this post===

1.00g of NiCl2.6H2O in 25.5ml of water with ethylenediammnie (1:1 molar)

Addition of Ni(en) solution to a mixture of KI solution with Pb(AcO)2 (4+:1 molar)

Bisethylenediamminenickel(II) bistri-iodoplumbate(II)
|
|
|
| Pages:
1
2
3
4 |









