UnintentionalChaos
International Hazard

Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Preparation of Styrene and Phenylacetylene from Polystyrene
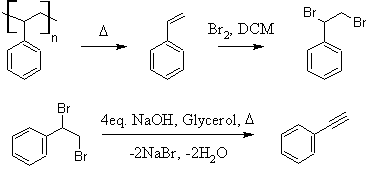
Introduction:
Phenylacetylene is a flexible organic intermediate with a myriad of potential uses. As a terminal alkyne, it undergoes many of the
characteristic reactions undergone by acetylene, but with the convenience of being an easily measured liquid. Terminal acetylenes are weakly acidic
and thus, it can be deprotonated by strong bases such as NaNH2 or Grignard reagents for use a nucleophile, such as in the preparation of rubrene (1).
Phenylacetylene can be hydrated using strong acids, mercury, gold, or other catalysts to give acetophenone or phenylacetaldehyde (2,3) It can be
homocoupled forming diphenyldiacetylene (4) or cross coupled with other alkynes via a Cadiot-Chodkiewicz coupling (5). Phenylacetylene is also
amenable to Sonogashira coupling.
Phenylacetylene has been prepared by a relatively small number of methods in the literature. Typically, either (1,2-dibromoethyl)benzene or
β-bromostyrene is treated with a strong base, inducing dehydrohalogenation(s) to form phenylacetylene. This can be a solution or melt of alkali
hydroxide (6,7) or a solution of sodium amide in anhydrous ammonia (8,9). Molten alkali hydroxide rapidly destroys glassware. Sodium amide is
difficult to prepare for the amateur, requiring sodium metal and liquid anhydrous ammonia. The latter is especially difficult to work with, and
dangerous without a fume hood. For this reason, a solution of alkali hydroxide was chosen for the production of phenylacetylene.
As an immediate precursor, (1,2-dibromoethyl)benzene can be easily prepared by simple addition of Br2 to styrene (10). β-bromostyrene can be
prepared from cinnamic acid by bromination and reflux with an alkali carbonate in aqueous acetone (11). The Takai olefination utilizing CrCl2 and
haloforms can also be used to make β-halostyrenes from benzaldehydes (12). As this reaction is very reagent intensive, it is not worthwhile for
making unsubstituted phenylacetylene but may be useful for substituted phenylacetylenes. Due to its simplicity and high yield, the bromination of
styrene was selected for this preparation.
Styrene can be prepared by a number of methods. Due to the great variety and frequent complexity of the preparations, most will not be discussed
here. Cinnamic acid can be decarboxylated to yield styrene, but the preparation takes a long time and yield is poor (13). Direct conversion to
β-bromostyrene as mentioned above would be a more efficient use of cinnamic acid. Styrene is present in significant amounts in easily available
products such as fiberglass resins (14). This can most likely be easily vacuum distilled from the other components of the resin. Another simple route
to styrene is the thermolysis of polystyrene (15). The polymer undergoes thermally induced bond homolysis and styrene distills over. Because the cost
of the reagents for this last preparation is negligible, it was selected for production of styrene.
Experimental:
Styrene
A 100ml RB flask (that the experimenter is not emotionally attached to) is charged with 52.08g (0.50mol) of polystyrene. A convenient source of
fairly pure and dense polymer is clear, colorless plastic cutlery. Pure polystyrene is brittle and wire cutters easily break the utensils into small
pieces. Tapping may be necessary to settle the polystyrene into the flask so that it will all fit. No headspace is necessary. The flask is set up for
simple distillation with an air condenser and stopper in the 3-way adapter where a thermometer adapter would usually go. A heat-proof clamp must be
used to hold the flask. Do not use keck clips. The receiving flask should be 100ml in volume or larger and submerged as much as possible in an ice
bath.
The distillation flask is heated directly with a flame. The polystyrene will begin to melt and a thick white fume will start distilling over. It
condenses with astounding efficiency considering the high temp and lack of a water cooled condenser. With continued heating, the polystyrene will form
a melt. Apply heat until nearly the entirety of the plastic has decomposed and distilled over. This will only take a few minutes. A very small amount
of dark residue remains in the distillation flask and is difficult to remove. As soon as cooled, rinse all parts of the apparatus with acetone or
toluene to remove styrene before it polymerizes. The distillate consists of a yellow mobile liquid. If you must stop at this stage, store the crude
product in the dark and as cold as possible. A small amount of hydroquinone or other polymerization inhibitor may be added.
The crude product is purified by setting up the flask with a stirbar for simple vacuum distillation with a water aspirator or roughly equivalent
vaccum source. Lower pressures will make the styrene difficult to condense efficiently. The styrene cannot be distilled at atmospheric pressure or
significant losses due to polymerization will occur. The condenser should be pumped with icewater and the receiving flask (250ml, in preparation for
the next step) should be immersed in an ice bath. The flask is heated with stirring in a hot water or air bath. Pure styrene distills over at 47-48°C
(32torr) as a clear, colorless mobile liquid. Distillation is ceased when the stillhead temperature exceeds 50°C, yielding 34.96g of styrene (67.1%
yield). If the styrene is to be stored at this point, a polymerization inhibitor must be added. It is preferable to make the styrene immediately
before use.
(1,2-dibromoethyl)benzene
The styrene from the previous step is diluted with 35ml of dichloromethane and chilled in an ice bath, or preferably a freezer. A seperatory funnel or
addition funnel is charged with solution consisting of 53.86g of bromine (1eq) in 35ml of dichloromethane. A stirbar is added to the styrene solution
and the funnel set up over it. Bromine is dripped in rapidly with stirring at first. When the solution becomes lukewarm, the addition rate is slowed
down and the reaction flask is immersed in an ice bath. Toward the end of the bromine addition, a yellow color will remain in solution. If addition is
stopped, it fades. When a pale orange color that does not fade on standing remains in the reaction flask, cease addition. A small amount of bromine
solution may remain. Distill or rotovap off the dichloromethane to yield crude (1,2-dibromoethyl)benzene. CAUTION: This material is a
fairly potent skin sensitizer. This may be used directly in the preparation of phenylacetylene.
If purification is desired, the crude product is broken up and dissolved in 600g of 67% aqueous isopropanol. Allow to cool to room temperature, then
chill in an ice bath. The mass of crystals that forms is stirred into a slurry and vaccum filtered. Air is drawn through the product to help dry it.
Removal of solvent is completed by placing in a dessicator over sulfuric acid until constant weight is achieved. The product is a fluffy, pearly white
powder weighing 73.17g (82.6% from styrene, 55.4% from polystyrene) and melting at 69-71°C (uncorrected).
Phenylacetylene
A 500ml, 2-neck RBF (WARNING: The flask and thermometer are superficially etched by this procedure) is charged with 250ml of
glycerol. A large stirbar is added. One neck is fitted with a thermometer adapter so the bulb is fully immersed in the glycerol. Strong heating is
applied to reduce the viscosity of the glycerol and stirring is begun. 40g (1mol) of NaOH is added. At roughly 125°C, the NaOH will be almost fully
dissolved forming a pale yellow-tan solution. 65.99g (250mmol) of (1,2-dibromoethyl)benzene is added and the flask set up for simple distillation.
Stirring must be efficient. The melted (1,2-dibromoethyl)benzene and intermediate β-bromostyrene are immiscible in the glycerol and good contact
between the phases is essential. At ~144°C, the reaction mixture begins boiling vigorously from phenylacetylene distilling. Two phases collect in
the receiving flask, clear droplets of water and a cloudy organic phase The temperature is increased gradually until it reaches 200°C. There should
be almost no distillate coming over at this point. Heating is ceased.
The distillate is transferred to a small seperatory funnel. The phases do not separate particularly well. The receiving flask is rinsed with 20ml of
diethyl ether, and this is added to the distillate. After mixing, the phases readily separate and the lower, aqueous layer is drained off. The organic
phase is rinsed with 30ml of distilled water, and the slightly cloudy organic phase transferred to an Erlenmeyer flask along with a small amount of
anhydrous MgSO4 to dry it. When the organic phase is clear, it is filtered through a fluted filter paper into a 100ml RBF. The Erlenmeyer is rinsed
with 5ml of ether and added to the filter paper. Once the solvent has stopped passing through, the filter paper and dessicant is rinsed with another
5ml of ether to prevent loss. A chaser solvent is added to the flask. 20g of diethylene glycol was used by the author. A stirbar is added and the
flask set up for vacuum fractionation. A controllable air bleed is a necessary addition.
The ether is removed first under reduced pressure. It shouldn’t condense, even at ice water temperature due to the reduced pressure. The next
fraction to come over is the phenylacetylene. The air bleed should be adjusted so this fraction comes over at ~80°C. When the phenylacetylene is
depleted, the stillhead temperature will rise rapidly. The vacuum should be shut off when this occurs, immediately halting the distillation. The
distillate is a clear, colorless liquid weighing 15.30g; 59.9% yield from (1,2-dibromoethyl)benzene (49.5% from styrene, 33.2% from polystyrene).
Unlike styrene, phenylacetylene is not terribly polymerization prone and does not need a stabilizer added. Store in the dark and cold.

References
1) User Brillantschwarz of Versuchschemie.de’s post at http://www.versuchschemie.de/topic,10433.html; based on Vogel's Textbook of Practical Organic Chemistry, Fifth Edition
2) Organic Syntheses, Vol. 83, p.55 (2006).
3) Synthesis, 2007, 1121-1150
4) Organic Syntheses, Coll. Vol. 5, p.517 (1973)
5) Synthesis, 2011, 1541-1546
6) Modified procedure from Organikum, post on Sciencemadness.org by Garage Chemist Thread ID=14935
7) Organic Syntheses, Coll. Vol. 1, p.438 (1941)
8) J. Am. Chem. Soc., 1934, 56 (10), pp 2120–2122
9) Organic Syntheses, Coll. Vol. 4, p.763 (1963)
10) Vogel, Arthur. A Text-Book of Practical Organic Chemistry, 3rd edition. Pg. 900
11) J. Chem. Educ., 1991, 68 (2), p 161
12) J. Am. Chem. Soc., 1986, 108 (23), pp 7408–7410
13) Organic Syntheses, Coll. Vol. 1, p.440 (1941)
14) www.jamestowndistributors.com/userportal/pdfs/MSDS/bondo/401... accessed 1/28/2012
15) The Chemical Educator, Vol. 8, No. 5, Published on Web 8/30/2003
[Edited on 1-29-12 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
plastics
Hazard to Others
Posts: 141
Registered: 6-11-2009
Member Is Offline
Mood: No Mood
|
|
Great post
I have also performed the third step (styrene dibromide/(1,2 dibromoethyl)benzene to phenylacetylene) using potassium hydroxide in methanol according
to:
Fiesselmann and Sasse, Chem. Ber., 89, 1775 (1956).

|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Quote: Originally posted by plastics  | Great post
I have also performed the third step (styrene dibromide/(1,2 dibromoethyl)benzene to phenylacetylene) using potassium hydroxide in methanol according
to:
Fiesselmann and Sasse, Chem. Ber., 89, 1775 (1956).
|
What did you use the phenylacetylene for? Some of my batch is destined for a small (<50mmol) prep of 9,10-bis(phenylethynyl)anthracene. I want to
try out the Sonogashira coupling. Or maybe I'll attempt rubrene. Either way, it's going to glow.
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
plastics
Hazard to Others
Posts: 141
Registered: 6-11-2009
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by UnintentionalChaos | | Quote: Originally posted by plastics | Great post
I have also performed the third step (styrene dibromide/(1,2 dibromoethyl)benzene to phenylacetylene) using potassium hydroxide in methanol according
to:
Fiesselmann and Sasse, Chem. Ber., 89, 1775 (1956).
|
What did you use the phenylacetylene for? Some of my batch is destined for a small (<50mmol) prep of 9,10-bis(phenylethynyl)anthracene. I want to
try out the Sonogashira coupling. Or maybe I'll attempt rubrene. Either way, it's going to glow. |
Great minds and all that ...
One batch was used to make said 9,10-bis(phenylethynyl)anthracene. I used example one of this patent using half quantities:
http://www.freepatentsonline.com/3911038.html
I'm surprised I got anything as I synthesised the lithium amide myself as well as the anthraquinone and phenylacetylene
The other batch I converted to 1,1,3-triphenylprop-2-yn-1-ol (Vogel 5th ed p539) ready for conversion to rubrene when I have time
I would also like to make some chloro substituted variants of the phenylethynlanthracenes by synthesising chloroanthraquinone via a Friedel-Crafts
condensation of phthalyl anhydride and chlorobenzene
|
|
|
chemx01
Hazard to Self
Posts: 54
Registered: 13-6-2010
Member Is Offline
Mood: No Mood
|
|
I made bis(phenylethynyl)anthracene with same procedure as plastic and I got a crappy yield, but nontheless I got about 2g of this anthracene which is
more than enough for my purposes.
I didn't use LiNH2 but I used LiH as a base to make lithium salt of phenylacetylene.
EDIT:
I finally put them on youtube so enjoy!
phenylacetylene: http://www.youtube.com/watch?v=aJkYgRIJDsA
anthracene: http://www.youtube.com/watch?v=kx9Ilfqp8RE
[Edited on 29-1-2012 by chemx01]
|
|
|
bbartlog
International Hazard
Posts: 1139
Registered: 27-8-2009
Location: Unmoored in time
Member Is Offline
Mood: No Mood
|
|
Nice writeup. I hadn't realized that heat alone could efficiently depolymerize polystyrene (I thought a catalyst e.g. sulfur was needed)... now I
know. Also surprised that phenylacetylene is so stable in storage.
The less you bet, the more you lose when you win.
|
|
|
Lambda-Eyde
National Hazard
Posts: 860
Registered: 20-11-2008
Location: Norway
Member Is Offline
Mood: Cleaved
|
|
So that's what you were going to use the dibromostyrene for! I didn't realize acetylenes could be prepared so easily - going from plastic cutlery to a
very interesting synthetic intermediate which opens for some even more interesting reactions! A very nice write-up, and I'm looking forward to seeing
the results of the coming syntheses utilizing the phenylacetylene. Will you post a video of the synthesis? Also, how's your synthetic capsaicin
project coming along?
| Quote: Originally posted by plastics | Great post
I have also performed the third step (styrene dibromide/(1,2 dibromoethyl)benzene to phenylacetylene) using potassium hydroxide in methanol according
to:
Fiesselmann and Sasse, Chem. Ber., 89, 1775 (1956).
|
What was your yield? It seems weird that people choose to carry out this reaction at 200 degrees and etching their precious flasks if it can be
carried out in refluxing methanol. Seeing as I am emotionally attached to most of my flasks, this route would be desirable... 
This just in: 95,5 % of the world population lives outside the USA
Please drop by our IRC channel: #sciencemadness @ irc.efnet.org
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
Nice synthesis, reminds me of some good times in my own lab several years ago. I used styrofoam as the polystyrene source and an old flat bottomed
24/40 flask for the pyrolysis reaction. I used a mixture of KOH and ethylene glycol for the dehydrohalogenation. I got a decent amount of
phenylacetylene, along with one of the partial dehydrohalogenation products. I have IR spectra of these compounds at my lab, but I am at graduate
school several hundred miles away currently. I think that I will have to make some more phenylacetylene since I still have about 60 mL of the styrene
from the pyrolysis reaction. I am thinking about making some cobalt tetraphenylcyclobutadiene complexes and I will need some phenylacetylene to couple
with bromobenzene via a Sonogashira reaction. I could also make some from trans-stilbene which I synthesized during Christmas break from benzaldehyde
via a cyanide catalyzed benzoin condensation followed by a Clemmensen Reduction, following some early Organic Syntheses procedures.
Amateur NMR spectroscopist
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
| Quote: Originally posted by benzylchloride1 | | Nice synthesis, reminds me of some good times in my own lab several years ago. I used styrofoam as the polystyrene source and an old flat bottomed
24/40 flask for the pyrolysis reaction. I used a mixture of KOH and ethylene glycol for the dehydrohalogenation. I got a decent amount of
phenylacetylene, along with one of the partial dehydrohalogenation products. I have IR spectra of these compounds at my lab, but I am at graduate
school several hundred miles away currently. I think that I will have to make some more phenylacetylene since I still have about 60 mL of the styrene
from the pyrolysis reaction. I am thinking about making some cobalt tetraphenylcyclobutadiene complexes and I will need some phenylacetylene to couple
with bromobenzene via a Sonogashira reaction. I could also make some from trans-stilbene which I synthesized during Christmas break from benzaldehyde
via a cyanide catalyzed benzoin condensation followed by a Clemmensen Reduction, following some early Organic Syntheses procedures.
|
That sounds fantastic. I am extremely jealous of your equipment, by the way.
I am not sure if you'd have any idea, but I posted a question related to this in the mechanism thread. On the double dehydrohalogenation of the
styrene dibromide. The first elimination (benzylic bromide) can go E1 or E2. The trans-beta-bromostyrene should be the dominant product, but this
lacks an anti-periplanar Br and H for the second dehydrohalogenation, which is not going to use E1, which would need a vinylic carbocation
intermediate. I would normally handwave and suggest a thermal isomerization, but the reaction works at liquid ammonia temperatures as well.
[Edited on 2-2-12 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
ThatchemistKid
Hazard to Others
Posts: 132
Registered: 2-6-2010
Member Is Offline
|
|
I preformed this scheme a few months ago, though I stopped at the dibromo compound, grad school now taking up my whole time and having me in a
different city than my lab is really puts a damper on things. But I found it useful to dissolve the polystyrene in acetone till the solvent was
saturated with the polymer then pour the resulting goop into the pyrolysis flask, much more styrene at once.
the acetone was removed prior to pyrolysis by just setting the flask in a hot water bath for a few hours while working on other things. Anything that
was trapped beyond that came over in the beginning of the heating for the pyrolysis step which I believe I actually did using a heating mantle not
flame.
excellent write up =D
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Here are some nice references that may lead to improved yield of phenylacetylene.
Also, in video form: http://youtu.be/P4FdbhWzvBE
A solid half of the video is discussion, both uses as mentioned above, mechanism, and improvements to the procedure to increase yield.
Attachment: 1241395.pdf (165kB)
This file has been downloaded 1871 times
Attachment: 1458858.pdf (207kB)
This file has been downloaded 3321 times
Attachment: cis-beta-bromostyrene.pdf (270kB)
This file has been downloaded 2143 times
[Edited on 2-4-12 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
Great,
I like to try it and make Rubrene by it.But in final step of making rubrene we need ultra vacuum!How we can provide it?what model of vacuum pump can
provide this pressure?
I have JB and Edward vacuum pump but they cant reach enough vacuum for this purpose
[Edited on 4-2-2012 by Waffles SS]
|
|
|
barley81
Hazard to Others
Posts: 481
Registered: 9-5-2011
Member Is Offline
Mood: No Mood
|
|
In the preparation of styrene, is it possible to distill the crude product at atmospheric pressure (with polymerization inhibitor)? Or, is vacuum
distillation required?
|
|
|
Dr.Q
Harmless
Posts: 24
Registered: 18-11-2012
Member Is Offline
Mood: No Mood
|
|
Great guide explains a lot. But i wonder something. In the Dibromostyrene step , i want to a substitution rather than a elimination with OH . How can
i do that to dibromo benzene ?
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
You don't. If you want the vic-diol, its best to hydroxylate the styrene. OsO4 is the "classic" reagent for this, but theres several others,
particularly as the terminal alkene means syn and anti dihydroxylation give the same product. I'm pretty sure theres a review somewhere of "Osmium
free dihydroxylation" reactions or something...
Found it: http://pubs.rsc.org/en/content/articlelanding/2011/cs/b92388...
Funny paper from someone that actually uses an aweful lot of Osmium! Very clever guy though.
[Edited on 18-9-2013 by DJF90]
|
|
|
Enkii
Harmless
Posts: 2
Registered: 14-2-2014
Member Is Offline
Mood: No Mood
|
|
would it be possible to use a poly-resin; styrene + maleic anhydride? if so how would one remove the maleic anhydride? and could one possibly retain
the MAA for future use?
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
| Quote: Originally posted by Enkii | | would it be possible to use a poly-resin; styrene + maleic anhydride? if so how would one remove the maleic anhydride? and could one possibly retain
the MAA for future use? |
Pol(Styrene-co-maleic anhydride) resin, you mean? I have no idea if the thermal depolymerization will work as smoothly as with pure nonpolar
polystyrene. I rather doubt that it will, but you could give it a shot. Probably, extraction of any free maleic anhydride by reaction to dissolve it
in aqueous solution is going to be your best bet.
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
subsecret
Hazard to Others
Posts: 424
Registered: 8-6-2013
Location: NW SC, USA
Member Is Offline
Mood: Human Sadness - Julian Casablancas & the Voidz
|
|
Sorry to revive an old thread...
Instead of Br2/DCM, could sodium hypochlorite solution be used to give chlorine instead of bromine?
Fear is what you get when caution wasn't enough.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I see no reason why chlorine won't work. Of course you need to acidify the hypochlorite with HCl and wait till all the chlorine moves to the top
(non-polar) layer.
Chlorine has solubility of 0.06 mole/L (2g/L) in HCl solution.
|
|
|
subsecret
Hazard to Others
Posts: 424
Registered: 8-6-2013
Location: NW SC, USA
Member Is Offline
Mood: Human Sadness - Julian Casablancas & the Voidz
|
|
I'm inclined to bubble chlorine straight into the styrene, maybe even styrene in DCM to make the chlorine more soluble. Chlorine is easier to work
with than bromine, in my opinion.
Fear is what you get when caution wasn't enough.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
actually you don't even need to make Cl2.why waste Cl2 or Br2 when you are going to rip it out in the next step.I
read recently that using a strong base(they used soda amide,but I think alc KOH should work),you can make acetylenes from halohydrins.
just react the styrene with HOCl(from bleach) and then react that with base to get phenylacetylene.
I was going to tell UC,but as this thread came up I am putting it here.
|
|
|
Oscilllator
National Hazard
Posts: 659
Registered: 8-10-2012
Location: The aqueous layer
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by byko3y | I see no reason why chlorine won't work. Of course you need to acidify the hypochlorite with HCl and wait till all the chlorine moves to the top
(non-polar) layer.
Chlorine has solubility of 0.06 mole/L (2g/L) in HCl solution. |
I tried this, and related my experience in a U2U I had with unintentionalChaos. Here's the relevant U2U:
| Quote: |
Well I performed the chlorination of styrene today, and had some strange observations. The first weird observation is that it appears that styrene is
(more or less) insoluble in dichloromethane, which is clearly inconsistent with your video (a problem on my part, I'm sure). I'll describe how I
obtained the DCM and styrene, so see if you can spot any potential reasons:
The dichloromethane I obtained yesterday from paint stripper that contained a significant (~15% IIRC) amount of methanol. This I removed with two
water washes, and I'm pretty sure the methanol was all removed because I measured the density of the water from the second wash, and it was almost
exactly 1g/cm^3. I then dried it over anhydrous MgSO4, then distilled it. The first part of the distillate came over slightly cloudy, so I think there
might have been a small amount of water left in the DCM.
The styrene was obtained in exactly the manner you outlined in your video only I used plastic shot glasses with recycling number 6 and a small "PS" on
the bottom instead of cutlery. I filled a shoddy 100ml flask twice, then distilled the resulting orange liquid under aspirator vacuum, collecting
everything boiling under 100 degrees. the distillate was slightly cloudy, though.
The chlorination went smoothly, with the only unusual observation being that the dischlorostyrene appeared slightly pink. I then went ahead to strip
off the DCM under vacuum, however I went a little to far, so a fair amount of the dichloromethane was removed. I did check the mixture periodically
though, and found to my immense surprise that the DCM and dichlorostyrene had swapped! the DCM now floated on top of the dichlorostyrene, as oppose to
the other way round.
The Dichloromethane is currently sitting in the freezer, but it refuses to solidify, instead oozing about like honey.
Any ideas what could have gone wrong? I might have another crack at it later on, on a larger scale...
|
|
|
|
DraconicAcid
International Hazard
Posts: 4412
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
"I did check the mixture periodically though, and found to my immense surprise that the DCM and dichlorostyrene had swapped! the DCM now floated on
top of the dichlorostyrene, as oppose to the other way round."
Wait- are you saying dichlorostyrene is not soluble in dichloromethane? That surprises me greatly.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
mackolol
Hazard to Others
Posts: 459
Registered: 26-10-2017
Member Is Offline
Mood: Funky
|
|
I'm having a little lack of understanding here.
First could phenylacetylene be deprotonated with lithium tert butoxide? Or is it too weak base (after all the lithium is connected with oxygen being
much more electronegative than hydrogen or nitrogen in amide). How do I check it? The pKa of the hydride and amide is way higher than that of tert
butoxide. Is it all about pKa?
And the second one. Could sodium phenylacetylide be replaced in bis phenylethynyl anthracene synthesis? I mean I'm sure that yields will go down as it
will be harder for the sodium rather than the lithium to be replaced, but would it yield anything?
I'm completely new to organometallic chemistry and am aware that electronegativity differences may not work analogical to organic halides here...
[Edited on 11-3-2022 by mackolol]
|
|
|








