mario840
Hazard to Others

Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
Reduction of oximes to corresponding amines
Hello
At first i wanna say that i tried reductive amination to prepare primary amines with of course 25% NH3 and yield was pathetic  , so i started search something more "yield" and i think that's reduction of oximes
, i found in some paper (references at the end) extremely easy preparation for example : acetoxime (withouth extraction , heating etc) look at this: , so i started search something more "yield" and i think that's reduction of oximes
, i found in some paper (references at the end) extremely easy preparation for example : acetoxime (withouth extraction , heating etc) look at this:
"Hydroxylamine hydrochloride (69g) was dissolved in 140 ml water and 140 ml 20% NaOH was added. Acetone (140 ml ) was added and the mixture was
vigorously stirred for 10 minutes. The reaction mixture was allowed to stand for one day and the crystals were collected by suction. Weight: 45 g or
62% yield."
Now we have our oxime so i wanna my amine in these case isopropylamine , they write the best for reduction is sodium and anhydrous ethanol (s....t
hell no ! too expensive) so i want to try Al/Hg reduction , i have make some changed recipe for that and i wanna know if that will works :
"We mix 125 ml methanol and 25 ml water and add to that in reaction flask 5 g al foil balls and 0,1 g HgCl2, we wait 20 minutes to finish amalgamation
, so we prepare second solution 11 g acetoxime in 50 ml methanol (should easily dissolve i searched and they write very soulible in alcohol), after 20
minutes we add our solution oxime to reaction flask and put reflux condenser , ater the reaction stops we chill to room temperature our solution and
now i don't know what to do :
1) extract with 2x50 ml toluene and gas with HCl
2) filtrate the solution and evaporate amine ?????
3) is that nessesary to add NaOH solution to dissolve remain Al foil ?
and most imporant question : did someone try reduction of oxime and how about yield ???? beacuse if is shitty , i give up
thnaks for any help
References :
C-13 Nuclear Magnetic Resonance Spectra of Amines by Cheng-Hsia Wang
|
|
|
arsphenamine
Hazard to Others
Posts: 236
Registered: 12-8-2010
Location: I smell horses, Maryland, USA
Member Is Offline
Mood: No Mood
|
|
several observations:
You can easily test all your work-ups on a 1/2 ml scale, and select the one you liike.
I'd make the acid adduct with HCl or citric acid in alcohol because i-PrNH2 stinks.
Consider running a Beckmann rearr. on the ketoxime to yield N-methyl acetamide ... which does NOT stink.
|
|
|
Methansaeuretier
Harmless
Posts: 45
Registered: 24-5-2010
Location: Europe
Member Is Offline
Mood: strongly alkaline
|
|
Offtopic, but does that may be an easy way to methylamine, by saponiffication of N-methyl acetamide?
Quote: Originally posted by mario840  |
1) extract with 2x50 ml toluene and gas with HCl
2) filtrate the solution and evaporate amine ?????
3) is that nessesary to add NaOH solution to dissolve remain Al foil ?
|
1) Dry toluene before gasing. Dropping conc. H2SO4 in IPA or acetone may be easier.
2) Steam-Destillation into HCl should be better and faster.
3) You can also try decant or filter the solution first, to remove unreacted foil, but you need to add that much NaOH to liberate the 2-propylamine
and dissolve any precipiating Al(OH)3 (not filterable) as soluble [Al(OH)4]+
[Edited on 28-9-2010 by Methansaeuretier]
[Edited on 28-9-2010 by Methansaeuretier]
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
so the step when i add NaOH to liberate amine MUST BE DONE ? or can i right after when reaction is over extract with toluene ?
|
|
|
Methansaeuretier
Harmless
Posts: 45
Registered: 24-5-2010
Location: Europe
Member Is Offline
Mood: strongly alkaline
|
|
| Quote: Originally posted by mario840 | | so the step when i add NaOH to liberate amine MUST BE DONE ? or can i right after when reaction is over extract with toluene ?
|
First it's no easy to extract MeOH with toluene, because MeOH will dissolve the toluene if you don't add water and NaOH
It also has to be basic because 2-propylamine salts are insolouble in toluene.
| Quote: |
"We mix 125 ml methanol and 25 ml water and add to that in reaction flask 5 g al foil balls and 0,1 g HgCl2, we wait 20 minutes to finish amalgamation
, so we prepare second solution 11 g acetoxime in 50 ml methanol (should easily dissolve i searched and they write very soulible in alcohol), after 20
minutes we add our solution oxime to reaction flask and put reflux condenser , ater the reaction stops we chill to room temperature our solution and
now i don't know what to do : |
A problem in your procedure is a missing H+ donor.
You have to add some formic- or acetic acid to get the reaction working I think.
Here's some literature with Zinc dust instead of aluminium amalgam, but may work similar:
| Quote: |
JOC 57, 6324 (1992)
At room temperature with stirring, Zinc dust (74 mg) was added to a solution of the oxime (44mg, 0.185 mmol) in 2 ml glacial acetic acid. Stirring was
continued for another 15 minutes. The reaction mixture was then filtered through a sintered glass funnel with suction. The filtrate was concentrated
under vacuum to afford the amine as an oil (37mg, 0.166 mmol, 90% yield). |
Correct workup is missing here I think... Zn-acetate isn't removed and the amine-acetate is produced, not the free amine.
| Quote: |
K. Abiraj and D. Channe Gowda
J. Chem. Res. (S) 6, 332-334 (2003)
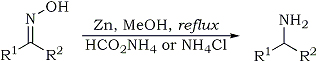
Reduction of oximes to amines, general procedure: To a solution of the substrate (5 mmole) in methanol (10 ml) was added ammonium formate (10-20
mmole) [or ammonium chloride (10-20 mmol)] and zinc dust (10 mmol). The mixture was stirred under reflux. After the completion of the reaction
(monitored by TLC), the reaction mixture was filtered through celite. The filtrate was evaporated under vacuum and the residue was taken into
chloroform or ether, washed twice with 80% saturated brine solution and finally with water. The organic layer was dried over anhydrous sodium sulphate
and evaporation of the organic layer was followed by purification either by preparative TLC, or by column chromatography, to yield the desired
product. |
In this case formic or acetic acid may work even better.
| Quote: |
K. Abiraj; D. Channe Gowda
Synthetic Communications 34(4), 599-605(2004)
To a solution of the substrate (10 mmol) in methanol or in any other suitable solvent (20 mmol) was added ammonium formate (30 mmol) and magnesium
powder (1g, 0.041 mol). The mixture was stirred under nitrogen atmosphere at room temperature. The reaction was exothermic and effervescent. After the
completion of reaction (monitored by TLC), the reaction mixture was filtered through celite. The organic layer is evaporated and the residue was
dissolved in chloroform or dichloromethane or ether and washed with saturated sodium chloride solution to remove excess ammonium formate. The organic
layer was dried over anhydrous sodium sulphate and evaporation of the organic layer followed by purification either by preparative TLC or by column
chromatography to yield the desired product. |
In this case formic or acetic acid may also work even better.
[Edited on 29-9-2010 by Methansaeuretier]
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
| Quote: |
A problem in your procedure is a missing H+ donor.
You have to add some formic- or acetic acid to get the reaction working I think.
|
I don't agree with that , why? look at in rhodium for reductiuon of oximes with al-hg , I want al-hg (i don't have acess to ammonium formate,
magnesium etc.):
"JACS 111, 6228 (1989) To a solution of the oxime (24.6 mg, 0.521 mmol) in 10% H2O-THF (30ml) were added aluminum amalgam, which was prepared from
0.25g of granular aluminum and 2% HgCl2 (0.1ml) in 10% H2O-THF (0.5ml). The reaction mixture was stirred for 3h at room temperature. 3ml conc NaHCO3
was added, and the reaction mixture was filtered and the filter cake washed with ethyl acetate. The filtrate was diluted with water and extracted with
ethyl acetate. The extracts was washed with brine, dried over MgSO4 and concentrated in vacuo to give 20.34 mg (85%) of the desired amine as an
oil."
As you know to amalagmation start there need to be water, so here it is 10% H2O-THF , and there is no mention to H+ donor like acetic acid or formic
(in other examples also don't add any of acid like formic or acetic) , sure there is NaOH in these case NaHCO3 witch is base, there only extract, wash
with brine and evaporate in vacuo and look at the yield ---> 85%
I think instead MeOH better will be to use Isopropanol wich form nice 2 layer after reduction, bottom - unreacted al and top aimne + some isopropanol,
add NaOH and extract with toluene or ether or etc.
By the way thx for reply , i wanna know if someone reduce oximes (not exactly these one) what yield he got 
|
|
|
Methansaeuretier
Harmless
Posts: 45
Registered: 24-5-2010
Location: Europe
Member Is Offline
Mood: strongly alkaline
|
|
| Quote: Originally posted by mario840 | | Quote: |
A problem in your procedure is a missing H+ donor.
You have to add some formic- or acetic acid to get the reaction working I think.
|
I don't agree with that , why? look at in rhodium for reductiuon of oximes with al-hg , I want al-hg (i don't have acess to ammonium formate,
magnesium etc.):
"JACS 111, 6228 (1989) To a solution of the oxime (24.6 mg, 0.521 mmol) in 10% H2O-THF (30ml) were added aluminum amalgam, which was prepared from
0.25g of granular aluminum and 2% HgCl2 (0.1ml) in 10% H2O-THF (0.5ml). The reaction mixture was stirred for 3h at room temperature. 3ml conc NaHCO3
was added, and the reaction mixture was filtered and the filter cake washed with ethyl acetate. The filtrate was diluted with water and extracted with
ethyl acetate. The extracts was washed with brine, dried over MgSO4 and concentrated in vacuo to give 20.34 mg (85%) of the desired amine as an
oil."
As you know to amalagmation start there need to be water, so here it is 10% H2O-THF , and there is no mention to H+ donor like acetic acid or formic
(in other examples also don't add any of acid like formic or acetic) , sure there is NaOH in these case NaHCO3 witch is base, there only extract, wash
with brine and evaporate in vacuo and look at the yield ---> 85%
I think instead MeOH better will be to use Isopropanol wich form nice 2 layer after reduction, bottom - unreacted al and top aimne + some isopropanol,
add NaOH and extract with toluene or ether or etc.
By the way thx for reply , i wanna know if someone reduce oximes (not exactly these one) what yield he got |
Aaah, H2O is the H-donor in that reaction I think.
Some hydrogen is absolutely neccessary! See picture!
H2O is a much better H-donor than MeOH and any acid should be a much better donor than water. But you can try it with water.

[Edited on 1-10-2010 by Methansaeuretier]
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
thx for reply , i think water and alcohol without acid will be fine , in my original recepture i thought MeOH will be good (of course it is) but
better to use isopropanol , that alcohol provide better extract of isopropylamine ! and definitely i will make sulphate salt , gassing or oparate with
fuming HCl is waste of time
|
|
|
Methansaeuretier
Harmless
Posts: 45
Registered: 24-5-2010
Location: Europe
Member Is Offline
Mood: strongly alkaline
|
|
You could add some aq. acetic acid after "your reaction" is over. It may increase the yield. Don't forget to add NaOH to liberate the amine. Na2CO3
does not work very good, because Al2(CO3)3 is not easy to filtrate.
|
|
|
scrambles
Harmless
Posts: 4
Registered: 20-10-2011
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Methansaeuretier |
| Quote: |
K. Abiraj and D. Channe Gowda
J. Chem. Res. (S) 6, 332-334 (2003)
Reduction of oximes to amines, general procedure: To a solution of the substrate (5 mmole) in methanol (10 ml) was added ammonium formate (10-20
mmole) [or ammonium chloride (10-20 mmol)] and zinc dust (10 mmol). The mixture was stirred under reflux. After the completion of the reaction
(monitored by TLC), the reaction mixture was filtered through celite. The filtrate was evaporated under vacuum and the residue was taken into
chloroform or ether, washed twice with 80% saturated brine solution and finally with water. The organic layer was dried over anhydrous sodium sulphate
and evaporation of the organic layer was followed by purification either by preparative TLC, or by column chromatography, to yield the desired
product. |
In this case formic or acetic acid may work even better.
[Edited on 29-9-2010 by Methansaeuretier] |
A lot of literature seems to indicate that this is a viable pathway for the reduction of oximes to their corresponding amines.
One of the authors of this paper was previously a professor in India and now is at Penn State. I'm considering sending him an email to see if he has
any proposed reaction mechanisms for this reaction.
|
|
|
fledarmus
Hazard to Others
Posts: 187
Registered: 23-6-2011
Member Is Offline
Mood: No Mood
|
|
@scrambles - this appears to be a typical catalytic metal reduction, using Zn as the catalytic surface and ammonium formate or ammonium chloride as
the hydrogen source. If you look up zinc ammonium formate on google scholar, you will find a lot of reductions of nitrogen in almost any oxidation
state down to the amine. Nitro, nitroso, oxime, imine, etc.
A very useful reaction and one of the reasons I keep a bottle of zinc dust under my lab bench! 
|
|
|
kmno4
International Hazard
Posts: 1503
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by fledarmus | this appears to be a typical catalytic metal reduction, using Zn as the catalytic surface and ammonium formate or ammonium chloride as the hydrogen
source. If you look up zinc ammonium formate on google scholar, you will find a lot of reductions of nitrogen in almost any oxidation state down to
the amine. Nitro, nitroso, oxime, imine, etc.
|
Burn your Zn, you need illumination.
I see that you have never conducted such reduction. Better to say nothing than rubbish.
Zn is not a catalyst in this reduction - it is one of reagents (->ZnO). In such reductions by metals + H-donors, mechanism is
always complicated.
|
|
|
scrambles
Harmless
Posts: 4
Registered: 20-10-2011
Member Is Offline
Mood: No Mood
|
|
The workup is very poorly described in the Zinc/Ammonium Formate reaction, and the author failed to say that any amine created would exist as the
formate salt, if I'm not mistaken.
|
|
|
scrambles
Harmless
Posts: 4
Registered: 20-10-2011
Member Is Offline
Mood: No Mood
|
|
Oh, nevermind that last post. Sorry, I got confused and was thinking formic acid was used rather than the ammonium salt.
Anyways, kmno4 says that ZnO is the reagent in this synthesis. I'm sorry, I am not too familiar with this reaction, let alone the mechanism, but from
my understanding, I thought it proceeded in the following way:
1) Ammonium Formate dissolved in water produces acidic solution, protonates subrate N
2) Zn donates electron to Carbon atom
3) Substrate N further protonated
4) Zn donates another electron to Carbon atom, releasing water molecule
5) freebase amine + Zn(COOH)2 + H20
But if ZnO were the primary reagent in this synthesis, it would be an oxidation then, correct? Oxidation would lead to regeneration of the ketone,
and one of the major uses of oximes is for the long-term storage of ketones. I'm not interested in recovering the ketone, this synthesis is supposed
to be a route to an amine.
EDIT:
Also, with the production of hydrogen from zinc + acid, perhaps this is a hydrogenation rather than a reduction? Perhaps a side reaction is ketoxime
to hydroxylamine?
[Edited on 24-10-2011 by scrambles]
|
|
|
fledarmus
Hazard to Others
Posts: 187
Registered: 23-6-2011
Member Is Offline
Mood: No Mood
|
|
@kmno4;
Thank you for your no-doubt well-intentioned critique of my post. Since the literature prep that I keep on hand for running these reactions (Gowda, et
al, Ind J Chem B (2001) p75-77 referred to these reactions as "zinc-catalyzed" and since the inorganic salts were filtered off at the end of the
reaction, it never occurred to me to look further into the mechanism than to note its similarity to the Pt/C:ammonium formate reductions. However, as
the Zn and Ni versions of the reaction do use a more-than-stoichiometric amount of metal, I would be tempted to accept your description of metal
oxidation as part of the mechanism.
You wouldn't happen to have a source that more fully explains the mechanism of such metal-"catalyzed" ammonium formate/acetate/hydrazine reductions,
would you? Not that I doubt your understanding, but in the 20+ years that I have been working in organic chemistry as a professional medicinal
chemist, I have come to rely more on peer-reviewed journal articles than word-of-mouth.
And I believe I will keep my Zn dust in its usual, useful place beneath my lab bench and in my battery of reductive techniques for now. I find
sufficient illumination in my occasional forays on chemical forums for my present purposes.
|
|
|








