| Pages:
1
2 |
Mephisto
Chemicus Diabolicus

Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
Dimethylsulfate - synthesis
I want to know your opinion how it could be possible to improve this synthesis. I have tried a DMS synthesis based on Rhodium's diethyl sulfate
synthesis, but get only a very small yield. Here's exactly what I have done:
[color=darkred]500 grams of sodium sulfate were placed in a dry 4 liter flask connected with a condenser and a receiver arranged for vacuum
distillation (see below). The flask was heated by means of an oil bath to 150 °C. The apparatus was exhausted as nearly as possible by means of a
filter pump, and a mixture of 200 grams of methanol and 590 grams of concentrated sulfuric acid was allowed to drop on the sodium sulfate. The
distillation of the mixture required about one hour. The part of the mixture, which didn't distil over at 150 °C remained in the 4 liter flask.
The DMS was separated from the top layer with a separatory funnel. It was washed with a dilute solution of sodium carbonate and then once with cold
water, then dried with anhydrous sodium sulfate. Yielding only 30 ml of DMS.[/color]
The set-up:
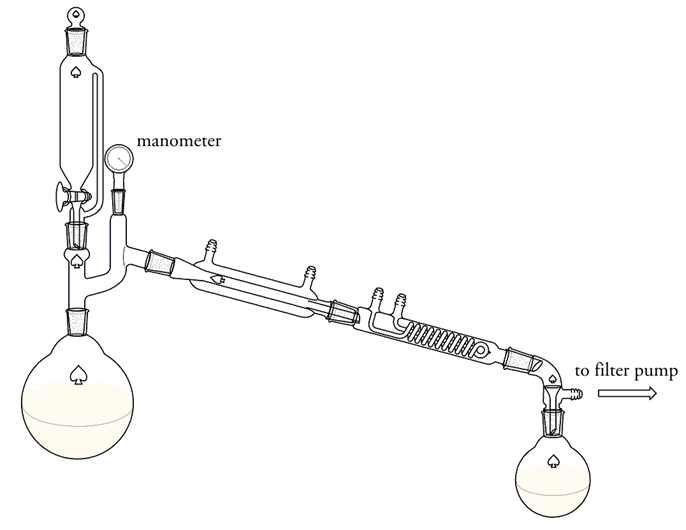
What did I wrong? I thought the yield would be at least 120 grams. Should I distil at a higher temperature, so that the whole mixture could
distil over?
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
And some information material
If someone wants to give the DMS synthesis a try, he should read at first the Dupont DMS Bulletin. Here.
The article about dialkyl sulfates from Ullmanns is also very interesting. Exported pages as PDF on the FTP (if the whole Ullmann is to big for
you ). Ullmanns - dimethyl sulfate.pdf ). Ullmanns - dimethyl sulfate.pdf
And in Houben-Weyl you can find some interesting informations as well. I scanned them, but only a German version of the text (64 pages; 1,27
MB). It's on the FTP.Pages from Houben-Weyl (German) - Methoden zur Herstellung und Umwandlung von Estern der Sauerstoffsäuren des
Schwefels.pdf
|
|
|
DDTea
National Hazard
Posts: 940
Registered: 25-2-2003
Location: Freedomland
Member Is Offline
Mood: Degenerate
|
|
I think you might be interested in the section about DMS from War Gases. The method they use is Ullmann's method again, but they have some
interesting references:
"Guyot and Simon, employing oleum containing 60% SO3 in the last method [the SO3 being reacted with Methanol] have obtained a very high yield of
Dimethyl Sulfate (about 90%)."
However, as you've probably found by your reading, the method preferred to produce DMS is the Chlorosulphonic Acid route.
|
|
|
BromicAcid
International Hazard
Posts: 3253
Registered: 13-7-2003
Location: Wisconsin
Member Is Offline
Mood: Rock n' Roll
|
|
Do you remember the fuming body incident with Gloria Ramirez? You can do a google search for her name plus DMSO and get some hits if you're
interested but the point is, one of the theories for her body possibly producing corpious amounts of poison gas is that she had applied a topical
ointment containing large amounts of DMSO to her body, in the ambulance they administered oxygen in excess and the theory states that the DMSO in her
body could have added a few oxygens to produce dimethyl sulfate with the excess of oxygen. Could be a possible route to your dimethyl sulfate, even
if it isn't it still makes for an interesting read if a little far-fetched.
http://www.gasdetection.com/MDS/m092198.html
http://www.ga.k12.pa.us/curtech/physiolo/Avi/Ramirez.htm
http://home.earthlink.net/~hdcr/Fuming.htm
|
|
|
DDTea
National Hazard
Posts: 940
Registered: 25-2-2003
Location: Freedomland
Member Is Offline
Mood: Degenerate
|
|
That seems like an interesting route... There are a few websites that sell DMSO, and Oxygen is readily available at hardware stores for welding. The
temperatures required here are not that high-- just body temperature, and then cooling to allow for the crystallization of DMS. They say nothing of
yields, however..
|
|
|
BromicAcid
International Hazard
Posts: 3253
Registered: 13-7-2003
Location: Wisconsin
Member Is Offline
Mood: Rock n' Roll
|
|
| Quote: |
Oxygen is readily available at hardware stores for welding
|
Jeeze, around here oxygen might be readily availible from hardware stores, but it costs $7 for 18g!!!! NO ONE IN THEIR RIGHT MIND PAYS ALMOST $2 A
GRAM FOR OXYGEN!! (Yes, I used all caps, I feel strongly on the issue, okay?) But who knows, it might be
cheaper where you come from, personally I prefer the catalytic decomposition of H2O2 @ 35% fast and very very cheap.
I however am sceptical of this reaction taking place in the body of a human being at anywhere near an appreciable rate (i.e. I don't think it
really happened the way they make it sound) But who knows, it might work on the lab scale, although the oxidation seems a bit off, putting oxygen
between the methyl groups and the sulfur and all.
|
|
|
Haggis
Hazard to Others
Posts: 238
Registered: 1-12-2002
Location: Mid-America.
Member Is Offline
Mood: Lacrymating
|
|
Dimethyl sulfate...hmm. It's rapidly absorbed into the skin. Once inside the skin, the hydrolysis products are methanol and sulfuric acid.
Sounds like a wonderful way to die. You go blind as the methanol is attacking your optic nerve and liver, while the sulfuric acid is scarring your
veins, causing inflammation and possibly internal bleeding. Fantastic stuff. As simple as the synthesis is, be careful, this one can bite you back.
Great ideas often receive violent opposition from mediocre minds.
<b> <a href=\"http://pgp.mit.edu:11371/pks/lookup?op=get&search=0xEE41A2B1\">PGP Key</a> </b> 0C0A 7486 B97F
92EE AE50 A98C A4F3 087E 8CE9 A782
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
The 'DMSO to DMS way' is interesting. But I'm only interested in a synthesis with methanol as starting material.
Samosa: I already thought about the synthesis with oleum and methanol from "The War Gases". But I only found an expensive oleum-source.
It's possible to make your own oleum this way:
(1) 3 FeSO<sub>4</sub> x aq --[O<sub>2</sub>;<font face="symbol">D</font>]--> FeO +
Fe<sub>2</sub>(SO<sub>4</sub><sub>3</sub>
(2) Fe<sub>2</sub>(SO<sub>4</sub><sub>3</sub>
--> Fe<sub>2</sub>O<sub>3</sub> + 3 SO<sub>3</sub>
(3) SO<sub>3</sub> + H<sub>2</sub>SO<sub>4</sub> --> oleum
However, for me this is to complicated and time-consuming as for a synthesis of a basic material.
Never thought about the way with chlorosulphonic acid, because this chemical isn't easy to get, too.
Haggis: I know this. I used a ABEK-standard-gasmask and worked in a good ventilated area.
Maybe the yield of my synthesis above could be improved by changing some parameters? What do you think?
|
|
|
Marvin
National Hazard
Posts: 995
Registered: 13-10-2002
Member Is Offline
Mood: No Mood
|
|
Was the 500g of sodium sulphate anhydrous, it isnt clear from the description or on Rhodium. I think it should be. How much water in the methanol,
shouldnt be too much normally, how much is in the sulphuric acid though?
Aside from these I would be worried that the sulphuric will soak up any water vapour in the 'vacuum', if the filter pump is the water type
this could be a serious problem. Also to check, the vacuum was isolated during the actual distillation?
I might also be inclined to try less sodium sulphate and drop the mixture in over a longer period, but this is a 'tweak' it wont affect
things enough either way.
Ive heard scare stories about how toxic DMS is, I dont plan on using it in future. If I need a methylating agent I might use sodium methyl sulphate.
This isnt volatile and sounds easier to make with 'home' concentration sulphuric. Would this work for your application?
Edit in reply to last to posts.
Haggis, nonononononooooo!
Its worse, its a methylating agent which makes it much more toxic than its constituents. Like 20 times, in addition to being carcinogenic and a whole
bunch of other things that go hand in hand with methylating agents.
Mephisto, the synthesis doesnt need an 'improved yeild', its failing at somepoint and you need to track down why. Work out what is being
done wrong, and get a decent yeild from it. Alternativly, use a different methylating agent for the problem.
[Edited on 2-3-2004 by Marvin]
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
I'm just passing on second-hand information here, but recently there was a short discussion about preparing DMS on the Hive itself.
Vitus_Verdegast, I believe, suggested that it was better to mix the methanol, H2SO4, and Na2SO4 for some time, then decant the liquid and distill it.
The idea was that with that method, any water bound by the Na2SO4 couldn't un-bind when the temperature was raised. I don't know if this
modified method had been tested.
PGP Key and corresponding e-mail address
|
|
|
DDTea
National Hazard
Posts: 940
Registered: 25-2-2003
Location: Freedomland
Member Is Offline
Mood: Degenerate
|
|
| Quote: | | Ive heard scare stories about how toxic DMS is, I dont plan on using it in future. If I need a methylating agent I might use sodium methyl sulphate.
This isnt volatile and sounds easier to make with 'home' concentration sulphuric. Would this work for your application?
|
This is of interest, because if you can make Sodium Methyl Sulfate, perhaps you could make Methyl Sulfuric Acid from this...which, on heating, forms
Dimethyl Sulfate.
With proper care, DMS is a wonderful Methylating agent, and can act as a cheaper substitute for Methyl Iodide. That's all Samosa has to say on
the issue of safety 
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
Marvin: The sodium sulfate was absolute anhydrous. I dried it 2 hours at 240 °C. The methanol was OTC-bought - I don't know how anhydrous it
was. H2SO4 was 96%. I never heard, that the water vapour from the filter pump (yes it was the water type) could get to the distilling-flask. For this
it had to pass through both condensers. At 10 mbar it would condense there. The whole apparatus wasn't very well isolated. Therefore the filter
pump ran the whole time, to prevent a higher pressure than 10 mbar during the synthesis. So a low air-stream passed through the set-up (from the not
airtight fittings to the filter pump). The humidity of this air-stream could be a problem, but I think that's of almost no consequence.
I think the fault was not to distil above 150 °C. There was a big rest of the MeOH,H2SO4,Na2SO4-mixture, which won't distill over at 150 °C. I
think the yield would be better if I would distill to 180 °C - even if the part of by-products gets higher.
Polverone: I will check this. Till now I only read half-apint's method for DMS at the hive (which seems to be good, but isn't proved yet).
|
|
|
DDTea
National Hazard
Posts: 940
Registered: 25-2-2003
Location: Freedomland
Member Is Offline
Mood: Degenerate
|
|
Did you try drying your OTC Methanol with a suitable desiccant? Also, you might want to try boiling the H2SO4 until it begins to fume--I'm pretty
sure this is important for the reaction.
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
Next time I will dehydrate the methanol with some CaCl2 and boil off the water from the H2SO4. I think the possible 99% methanol from the
CaCl2-dehydration will be enough and boiling with some magnesium powder under reflux won't be necessary.
|
|
|
Marvin
National Hazard
Posts: 995
Registered: 13-10-2002
Member Is Offline
Mood: No Mood
|
|
No dont dehydrate the methanol with CaCl2 they are incompatable.
Commercial methanol should be fairly anhydrous, I'd be more worried about the acid.
SciGuy and Vitus_Verdegast have the idea that dehydration is performed by the sodium sulphate removing the water from the reaction, Verdegast then
goes furthur to suggest that dehydration should be done in the cold, followed by decantation and distillation to avoid the heat decomposing the
fragile hydrated sodium sulphate. This is presumbly founded in the use of sodium sulphate to dry organic extractions. This doesnt sound right to me
as sulphuric acid itself is a much stronger dehydrating agent and what we know doesnt fit his ideas. We can form the monomethyl sulphate in decent
yeild mearly by boiling sulphuric acid with methanol (or ethanol) in a 2:1 by weight ratio, letting cool and neutralising. (fownes, but similar to a
rhodium synth in the nitroalkane section)
It seems more likley to me the use of the sodium sulphate is in some way related to avoiding the production of diethyl ether or ethene. e.g. sodium
methyl sulphate maybe decomposing to dimethyl sulphate and sodium sulphate at heat.
Just when I thought my entire book collection contained only a single paragraph on dimethyl sulphate (telling me how great it is for methylations due
to its high boiling point), fownes turns up a prep for methyl hydrogen sulphate (above), similar to the rhodium one, and a surprisingly simple prep
for dimethyl sulphate :- 8 to 10 parts of conc sulphuric acid mixed with 1 part of methyl alcohol and distilled neerly to dryness. The layers of the
distillate are seperated and the DMS neutralised and dried. The same book also has a method for diethyl sulphate and this is *not* analagous, it
involves passing sulphur trioxide into diethyl ether. The reason presumably is that ethyl alcohol will dehydrate to ethene under those conditions
(150C or so, sulphuric in excess), whereas methyl alcohol cannot.
I think based on this there are some plausable variations you can try, and be aware that dimethyl sulphate is supposed to decompose at its atmospheric
pressure boiling point, the main reason for the vacuum. Fownes is an old reference (middle 19th century) but it does seem reliable. This said its
methods are frequently not optimal. SciGuy suggests molar ratios of methanol to acid in his (only!) post on this forum, and also the use of magnesium
sulphate, Verdegast on the hive also suggests that trimethyl phosphate can sometimes be used to replace dimethyl sulphate in methylations.
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
"parts"
Sorry, a bit unclear: 8 to 10 parts conc. H2SO4 to 1 of methanol means... moles? Grams? CCs? Also, is this an atmospheric pressure distillation in
Fownes? And does distilling nearly to dryness mean (as it seems) to distill the majority of the conc. acid as well?!
I like the utter simplicity of this method. If I were to try to come up with a DMS synthesis on my own, I would've said "use methanol and a
lot of H2SO4." It's nice to know that intuition can be right once in a while.
Edit: while searching for more information on Fownes, I found a reference to a rather clever preparation of aqueous HCN that he apparently gave: KCN
is decomposed with tartaric acid in an alcohol/water mixture; sparingly soluble potassium hydrogen tartrate precipitates and can be easily removed. I
think this book may bear ordering, if there are other such interesting bits in it...
[Edited on 3-5-2004 by Polverone]
PGP Key and corresponding e-mail address
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
A drying agent is necessary, because water shifts the equilibrium to the left side of the first reaction (lowers the yield).
1) MeOH + H2SO4 <---> MeHSO4 + H2O
2) 2 MeHSO4 <---> Me2SO4 + H2SO4
Mountain_Girl (hive) tried it without dehydration and got a very poor yield.
The claim of Vitus_Verdegast (to dehydrate the MeOH + H2SO4 -mixture and remove the hydrated sodium sulfate) is logically. Above 100 °C it loses
it's water again. My main fault was not to remove the Na2SO4 <b>·</b> 10 H20 from the mixture before distilling.
Marvin: I checked it in Roempp (chemistry lexicon): CaCl2 is right for methanol. Other possible desiccants are CaO, magnesium and 3A molecular sieves.
|
|
|
Marvin
National Hazard
Posts: 995
Registered: 13-10-2002
Member Is Offline
Mood: No Mood
|
|
Polverone,
Parts is usually by weight, particually in 19th century books as I'm sure youve seen before, yes I assume neerly to dryness means most of the
acid too. I agree, its a little strange. There is no mention of a vacuum, so I assume a improvement in the yeild is possible when you use lower
pressure. The sum of information so far implies that a yeild in excess of 50% is possible with methanol and sulphuric acid alone, based on methanol.
I am unable to find the HCN synth by that method in my copy. The index is shockingly bad, even considering the language. There are also a lot of
versions of this book and some may be much better than others. Since the lethal dose for an adult is something like 50mg of HCN, I wont be trying it.
It does bring to mind a similar synth where a weak acid is left almost pure in solution by ppting alum though. Some odd ideas and some very clever
ones. The plan is to scan the book when I get back to scanning, I only have volume 2 though (organic compounds).
Mephisto,
"Mountain_Girl (hive) tried it without dehydration and got a very poor yield."
Sulphuric acid is a very good dehydrating agent, so I dont share your concerns. What Mountain_Girl tried was a variation at low temperature, high
concentration of alcohol and with the DMS voltalised with a stream of gas based on a british patent. Water was also sucked back into the reciver from
the trap. From the description it is unsurprising it failed.
I did make a mistake with the sodium sulphate. What can I say, my credibility is completely ruined  I assumed sulphuric was in excess and it isnt, serves me right for shirking the math. If sulphuric was in excess
this would be the stronger dehydrating agent, when not in excess its quite plausable sodium sulphate hydrates. I am under the impression that with
methanol the sodium sulphate method is less of an improvement then with ethanol, but it should help. Methanol forms esters more easily (rapidly,
completely) than ethanol. I assumed sulphuric was in excess and it isnt, serves me right for shirking the math. If sulphuric was in excess
this would be the stronger dehydrating agent, when not in excess its quite plausable sodium sulphate hydrates. I am under the impression that with
methanol the sodium sulphate method is less of an improvement then with ethanol, but it should help. Methanol forms esters more easily (rapidly,
completely) than ethanol.
"I checked it in Roempp (chemistry lexicon): CaCl2 is right for methanol."
CaCl2 cannot be used for drying alcohols or amines (including ammonia) becuase it forms adducts, acids (including phenols) and rather oddly esters.
This alcohol effect is sometimes used to purify methanol, you form the hex-adduct, evaporate to crystals and decompose the crystals with a large
amount of water, followed by distillation and dehydration. Calcium oxide, anhyd. sodium or magnesium sulphate would be fine for drying methanol. The
magnesium metal method is the best, but a bit extreme for the circumstances.
There is a lot of information on the hive, some genius, some utter stupidity and a lot inbetween. The method halfapint is using with alcohol,SO2 and
CuCl2 is interesting and I partulally like his method for generating SO2 from sulphuric acid and sulphur. The patent that uses sodium pyrosulphate
and alcohol alone looks particually promising (USP1506228) for a home synth.
|
|
|
BromicAcid
International Hazard
Posts: 3253
Registered: 13-7-2003
Location: Wisconsin
Member Is Offline
Mood: Rock n' Roll
|
|
How about a strong, possibly pyro phosphoric acid, that should dehydrate like no-one's business. But it might react under these conditions.
Well, anything to drive the equilibrium to the right, eh?
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
Marvin: Excellent work!
I am under the impression that with methanol the sodium sulphate method is less of an improvement then with ethanol, but it should help.
Yes, now I read in Houben-Weyl, that the Na2SO4 improves especially the reaction with ethanol (also NaCl could be used instead of the sulfate).
Fownes' method sounds not very scientific. Without vacuum most of the product would be destroyed. Also distilling to dryness will cause this: At
first the dimethyl sulfate comes to the receive-flask, then at higher temperatures the sulfuric acid. Both react back this way:
Me2SO4 + H2SO4 <---> 2 MeHSO4
BTW (Marvin): PM my if you need some help with OCR and PDF-creation from the originals. Some ideas in Fownes could be better than the DMS-method.
Next time I will use some more H2SO4 and Na2SO4 - if I'll use it at all - just in the cold.
|
|
|
thefips
Harmless
Posts: 33
Registered: 6-3-2004
Location: Germany
Member Is Offline
Mood: self destructive
|
|
Hi @ all! This is my first post.
I think that it is not very difficult to make oleum.
SO3 can be made by heating FeSO4 to ~400°C or mixing conc. H2SO4 with P2O5.
Perhaps 96% H2SO4 could be dried by mixing with Na2SO4 and separating the liquid phase,but I am not sure about this.
[Edited on 6-3-2004 by thefips]
|
|
|
BromicAcid
International Hazard
Posts: 3253
Registered: 13-7-2003
Location: Wisconsin
Member Is Offline
Mood: Rock n' Roll
|
|
| Quote: |
Yes, now I read in Houben-Weyl, that the Na2SO4 improves especially the reaction with ethanol (also NaCl could be used instead of the sulfate).
|
Are you saying to use NaCl in place of Na2SO4 with H2SO4 and MeOH? If that was the case you would probably end up with just HCl considering that
H2SO4 reacts with NaCl. If it somehow didn't you would end up with methyl chloride, same as the procedure to make methyl iodide.
Or am I misunderstanding?
Also thefips, your oleum production method is mentioned further up thread.
[Edited on 3/7/2004 by BromicAcid]
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
Are you saying to use NaCl in place of Na2SO4 with H2SO4 and MeOH?
No. I was talking about the analogue reaction, which ends up with Diethylsulfate. This reaction with EtOH is
improved by adding Na2SO4 or NaCl. Apparently the synthesis of DMS from MeOH and H2SO4 isn't improved that way.
The DES-synthesis improved by NaCl:
(Note: 2 moles ethyl hydrogensulfate are with 1 mole sodium chloride in one pot.)
C2H5OSO2OH + NaCl → C2H5OSO2ONa + HCl
C2H5OSO2ONa + C2H5OSO2OH → (C2H5)2SO4 + NaHSO4
The reaction could by improved because the ethyl hydrogensulfate would react further with the sulfuric acid (according to Houben-Weyl). Methyl
hydrogensulfate doesn't react further this way.
[Edited on 7-3-2004 by Mephisto]
|
|
|
BASF
Hazard to Others
Posts: 282
Registered: 5-11-2002
Member Is Offline
Mood: hydrophilic
|
|
Maybe this patent is useful too..
It describes the formation of sodium methyl sulfate using sodium sulfite, iodine(as an oxidation agent to form SO3 in situ) in aqueous (!) methanol.
The yield seems satisfactory
Description of DE4135728
Schwefeltrioxid, das Anhydrid der Schwefelsäure, ist eine seit langem bekannte und für Sulfurierungsreaktionen eingesetzte Verbindung. Es gibt
deshalb auch eine Reihe von Verfahren für seine Darstellung, die ausführlich aus anorganischer Sicht in Gmelins Handbuch der anorganischen Chemie 9
(Schwefelband) Teil A, S. 320- 484 und Teil B, S. 323-366 (8. Auflage, 1953), Verlag Chemie, Weinheim, beschrieben sind. Erinnert sei an das
Bleikammer- bzw. Kontaktverfahren. Bei beiden wird aus Schwefeldioxid und Sauerstoff im ersten Schritt Schwefeltrioxid hergestellt.
In der organischen Chemie wird Schwefeltrioxid meist als Reagenz eingesetzt, um organische Verbindungen mit beweglichem Wasserstoff wie Alkohole,
Phenole, aliphatische und aromatische Amine, aromatische Kohlenwasserstoffe oder auch Heterocyclen zu sulfurieren. Hierbei kann das Schwefeltrioxid je
nach Struktur der umgesetzten Substanz und den Reaktionsbedingungen auch an unterschiedlichen Stellen des Moleküls angreifen und eintreten. Bei der
Verwendung von Anilin als Modellsubstanz bildet sich bekanntlich mit Schwefeltrioxid erst Phenylsulfaminsäure, die sich bei erhöhter Temperatur in
Sulfaminsäure umwandelt.
An Stelle von SO3 können für Sulfurierungen auch Schwefeltrioxidderivate eingesetzt werden wie Schwefelsäure, Chlorsulfonsäure,
Chlorsulfonsäureester bzw. Schwefeltrioxid-Addukte an Pyridin, Dioxan oder Dimethylformamid.
Diese Verfahren und Methoden sind z. B. ausführlich beschrieben im Houben-Weyl, Methoden der organischen Chemie, Band 9, S. 343-599 (4. Auflage,
1955) bzw. im Band E11, S. 1042 ff (1985), Georg Thieme Verlag, Stuttgart. Eine eingehende Darstellung findet sich auch in dem Artikel von E.E.
Gilbert in Chem. Rev. 62, 549-589 (1962) unter der Überschrift: "The reactions of sulfur trioxide, and its adducts, with organic
compounds".
Wie aus den Beschreibungen hervorgeht, sind diese Verfahren nicht universell anwendbar. Es besteht deshalb Interesse an der Entwicklung weiterer, vor
allem preiswerter Verfahren.
Wir fanden jetzt überraschend, dass sich Schwefeltrioxid auch in wässrigem Milieu bildet, wenn man Sulfite mit Halogen umsetzt. Wir nehmen folgenden
Reaktionsweg an: Überraschend ist dieser Befund deshalb, weil nach bisherigem Lehrbuchwissen bei dieser Reaktion Schwefelsäure bzw. Sulfat entsteht.
Diese entstehen nach unseren Untersuchungen jedoch erst in einem 2. Schritt, in dem sich das Schwefeltrioxid mit Wasser umsetzt. Überraschend ist
weiter, dass das Schwefeltrioxid unter diesen Bedingungen nicht nur mit Wasser reagiert, sondern auch mit gleichzeitig anwesenden organischen
Verbindungen mit beweglichem Wasserstoff. Damit steht ein weiteres Sulfurierverfahren zur Verfügung.
Gegenstand der Erfindung ist ein Verfahren zur Darstellung von Schwefeltrioxid, dadurch gekennzeichnet, dass Sulfite im Beisein von Wasser mit Halogen
umgesetzt werden. Ein weiterer Gegenstand der Erfindung ist die Verwendung des hergestellten Schwefeltrioxids für Sulfurierungsreaktionen, dadurch
gekennzeichnet, dass die Umsetzung von Sulfit mit Halogen im Beisein von sulfurierbaren Verbindungen durchgeführt wird.
Als Sulfite kommen die Alkali- und Erdalkalisulfite in Frage, aber auch Sulfite, deren Kationen aus protonierten (bzw. quartärnisierten) organischen
Basen oder Aminen bestehen. Auch entsprechende Bi- bzw. Disulfite sind einsetzbar.
Bezüglich der Halogene liefern Chlor, Brom und Jod gute Ergebnisse. Die Brauchbarkeit von Fluor ist zweifelhaft. Dafür können
Interhalogenverbindungen wie Jodchlorid oder Jodbromid Verwendung finden.
Als besonderer Vorteil der Erfindung wird angesehen, dass als Ausgangsstoffe des erfindungsgemässen Verfahrens Abfallprodukte anderer chemischer
Reaktionen eingesetzt werden können, die so einer Entsorgung und zugleich einer Wiederverwendung zugeführt werden. Zu denken ist hierbei z. B. an
die Absorption von bei der Verbrennung von Erdöl oder Kohle entstehendem SO2 in Lauge oder Amin und Einsatz des so gebildeten Sulfits in einem
erfindungsgemässen Prozess.
Wie in der Einleitung beschrieben, gibt es sehr viele sulfurierbare Verbindungen. Die nachfolgenden Beispiele sollen das Verfahrensprinzip
verdeutlichen. Die Anwendungsbreite des Verfahrens lässt sich durch fachmännisches Kombinieren natürlich viel weiter ausschöpfen.
Beispiel 1
Darstellung von Natriummethylsulfat
In 100 ml Wasser wurden 12,6 g Natriumsulfit gelöst. Unter Rühren wurden 100 ml Methanol zugesetzt. Zu der jetzt stark trüben Lösung wurde unter
Eiskühlung eine Lösung von 25,4 g Iod in 250 ml Methanol zugetropft. Die Mischung wurde mit etwa 10 ml 32%iger Natronlauge neutralisiert. Die
Lösungsmittel wurden am Rotationsverdampfer abgezogen.
Der Rückstand wurde mit 500 ml Aceton verrührt, um das gebildete Natriumiodid zu entfernen. Der verbliebene Rückstand wurde abgesaugt und mit 250
ml Methanol verrührt. Vom unlöslichen Rest (Natriumsulfat) wurde abfiltriert. Das Lösungsmittel des Filtrats wurde verdampft. Es verblieben 8,6 g
Natriummethylsulfat=63% d. Th.
Identifizierung durch Vergleich des IR-Spektrums mit dem einer käuflichen Probe.
Beispiel 2
Darstellung von Cyclohexylammoniumcyclohexansulfamat
In einer Mischung von 117 g Cyclohexylamin und 70 g Wasser wurden nacheinander unter Eiskühlung und Rühren 6 g Schwefeldioxid und 5 g Chlor
gasförmig eingeleitet. Die Lösungsmittel wurden am Rotationsverdampfer abgezogen. Der Rückstand wurde mit 50 ml Wasser gut verrührt. Anschliessend
wurde abgesaugt, mit Aceton gewaschen und getrocknet.
Ausbeute: 17,7 g=677% d. Th.
Das IR-Spektrum der Substanz entsprach dem einer aus äquimolaren Mengen von Cyclohexylamin und Cyclohexansulfaminsäure hergestellten
Vergleichssubstanz.
|
|
|
mechem
Harmless
Posts: 32
Registered: 27-12-2007
Member Is Offline
Mood: No Mood
|
|
Dimethyl sulphate prep idea and question
Hello everyone
Before I start I must stress DMS is HIGHLY TOXIC and all preparations and use must be carried out in a fume cupboard followed by cleaning all
glassware with a methanol/ammonia solution. Remember this substance can affect you hours later so do your research first please.
So far I have not come across any literature on its preparation, but this method might work and I would be delighted to hear about any genuine
lab procedure or any quires/ideas you might have with my method. Maybe for example boiling methanol/H2SO4 using a Dean/Stk to remove the water but not
heating over 140C. ( DMS decomposes )
I have not tried this yet, but if one mixed equal molar quantities of methanol and conc. H2SO4 in a dropping funnel placed on top of a 3-neck R/B
flask set up for vacuum distillation. Then dripped the meth/H2SO4 at a few drops per second into the R/B flask. The R/B flask contains an excess of
anhy Na2SO4 preheated to 140 C ( might be a slight problem here with water absorption ). The receiver flask being placed in an ice bath to allow the
DMS formed not to hydrolyze back. The DMS can then be washed and separated first with cold sodium carbonate sol. then water and dried over anhy sodium
sulphate.
Edit by Chemoleo: Please search before making new threads next time!
[Edited on 28-12-2007 by chemoleo]
|
|
|
| Pages:
1
2 |










