| Pages:
1
2
3
..
5 |
Formula409
Hazard to Others

Posts: 129
Registered: 13-12-2008
Member Is Offline
Mood: No Mood
|
|
Possible OTC, 'safe' methylating agents
Greetings.
After reading the literature available on the Hive and the journals, it appears that methyl esters of carboxylic acids are able to function as
methylating agents with reasonable yield (/chemistrydiscourse/000463170.html). The author of the post specifically mentions a journal article which
shows that methyl, and other esters of oxalic acid alkylate aromatics quite well. This article is attached and a synthesis of the compound can be
found here: http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv2... . The main advantage of dimethyl oxalate over compounds such as DMS and TMP are
its easy availability and low toxicity, with it not having any large warnings on the MSDS and it being a powder, preventing one's DNA getting
methylated when one tries to move the compound into the reaction vessel.
ning then proposes that methyl esters of other carboxylic acids found around the home (citric, tartaric) should be able to perform the same function.
It appears that nobody else has attempted to study these as I am having a hard time just finding basic data regarding the properties of these
compounds let alone their methylating powers!
Does anybody have any information to contribute regarding this? I am all for giving it a go, but my knowledge of methylation and designing synthesises
is lacking. What would be a good, legal substrates to asses the effectiveness of each agent on?
It would be great if others could perhaps have a go at preparing methyl esters of both tartaric and citric acids, or even oxalic acid and seeing
whether they work! I will be able to attempt a synthesis of all three (with pics) using Fischer Esterification within the next few days if all goes
well.
Formula409.
[Edited on 13-12-2008 by Formula409]
[Edited on 13-12-2008 by Formula409]
Attachment: dimethyloxalate.pdf (684kB)
This file has been downloaded 4336 times
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
The oxalic acid methyl and ethyl esters work because the monoalkyl oxalate is almost a good leaving group. Compare the pKa1 of oxalic acid and that of
other common carboxylic acids like acetic or citric (the pKa of acids generally strongly correlates to their leaving group ability in
S<sub>N</sub>2 reactions. Though esters of weak acids like carbonates, formates, acetates and phthalates have been reported to alkylate
phenols at extreme conditions (only most simple phenolic substrates can thus be used), the mechanism was found to be different (not
S<sub>N</sub>2). Of these only dimethyl carbonate is useful for preparative reactions.
As far as oxalates as alkylating reagents and home chemistry, you can find an example of ethylation of a simple phenol (eugenol) using diethyl oxalate
and K2CO3 at the Hyperlab forum. I do not know if that procedure would work for phenols substituted with more sensitive functional groups as it is
done under quite harsh conditions. Obviously since the reaction is a simple S<sub>N</sub>2 substitution, the methyl oxalate should react
at considerably milder conditions since the reactivity of RX goes in the direction MeX > EtX > i-PrX (where X is a leaving group).
Often the difference in reactivity between MeX and EtX is of order of few magnitudes.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Although this obviously is a interesting subject, I think it is very limited in scope. Like Nicodem said, only very few, thermicly stable phenols have
been alkylated, most of the time in medium to mediocre yields, by very high temps. Under such conditions, even hydrogenosulfates seem as efficient and
easily made.
I think other esters have much more potential, trimethylphosphate can be regarded as safe (as a chemical can be), and quite efficient for phenols, can
be used around 80°C which is more than suitable for medium-sensitive phenol substrates.
Alkyl sulfonates are easily made at home, and seem very promising. Ullmann (the meber) reported very nice yields using ethane sulfonates. Tosyl
chloride, although a bit less easily accesable, is a very quick and easy way to tosylates, which prove to be very good O- or N-alkylating agents .
MeBr is very easily made, and can be used in a way that prevents any exposure. Yields reported by Painkilla seem to be very satisfying.
Feel free to report back any results you can obtain with alkyl oxalates, these esters can be regarded as reagents for other reactions too (formation
of b-keto esters here, here, here, and here.
(lots of other examples available)
It can also be used in a indole squeleton synthesis from o-nitrotoluene here.
I think there are far more possibilities in condensation reactions than in O-alkylations.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Nevertheless, dimethyl- or diethyloxalates are relatively common chemicals and some may have less troubles obtaining these than the more common
reagents. Of course, if you have to buy a methylation or ethylation reagent then you better buy an efficient one. I would also not bother with
esterifying oxalic acid just to prepare these oxalates, but it is always nice to have a wider spectrum of reagents that can be used by home chemists.
Too bad that harsh conditions are required, but a reflux in DMF and K2CO3 as base is actually something that several simple phenols can withstand
without troubles and I have to thank Formula409 for sharing the attached paper now that I finally read it.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Formula409
Hazard to Others
Posts: 129
Registered: 13-12-2008
Member Is Offline
Mood: No Mood
|
|
Thank you for your responses. I should be able to post up a synthesis tomorrow hopefully. Just a quick question, though, what purpose does the DMF
serve in the methylation reaction? Is it just used as a solvent that will not react with the agent as it cannot be further methylated? Can it be
substituted with DMSO? I will have to purchase some (long wait) if I cannot find a suitable substitute.
Formula409.
[Edited on 15-12-2008 by Formula409]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
DMF is a dipolar solvent which serves many purposes in the reaction (to solvate the K2CO3 in order for it to deprotonate the phenol;
S<sub>N</sub>2 reactions run faster in polar solvents; it is a solvent that does not solvate the nucleophile and thus makes it more
reactive...). DMF can be O-methylated thus forming the MeO-CH=N<sup>+</sup>Me<sub>2</sub> species which can also
O-methylate either the phenoxide or monomethyl oxalate anions (thus making this irrelevant). However, at such temperature using DMSO may not
be a good idea since it can also get O-methylated but to a species that decompose to formaldehyde and dimethylsulfide (this is the reason why
alkylations are generally not done in DMSO at temperatures higher than 100°C). Instead of DMF you can use any equivalent solvent: NMP, dimethyl- or
diethylacetamide, sulfolane, tetramethylurea (expensive!) and so on. You can also use a less polar solvent like diglyme or polyethyleneglycol. Or you
can just do the reaction solventless like in the example described at the Hyperlab forum (I could post a direct link, but I'm not sure their
administrator would like this, so find it yourself - it is in the " The eugenoloids /
Эвгенолоиды" thread which is currently on the 11th page of the forum).
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Formula409
Hazard to Others
Posts: 129
Registered: 13-12-2008
Member Is Offline
Mood: No Mood
|
|
Ok, have obtained NMP. Going to try methylating a phenolic benzaldehyde with dimethyl oxalate both solventless and in NMP which I get time. A TON of
my equipment broke yesterday which will prevent me doing anything until I purchase more when the suppliers re-open after Christmas.
Formula409.
[Edited on 10-19-2009 by Polverone]
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
Actually, CHO(and other EWG-groups) in o,p-positions to OH make it less active, and in case of such weak alkylators like dimethyl oxalate it may be
critical. If the experiment fails, try to prolong the time of reaction and/or increase the temperature.
|
|
|
detritus
Harmless
Posts: 19
Registered: 27-4-2008
Member Is Offline
Mood: No Mood
|
|
@Nicodem,
I looked but could not find the hyperlab article you mentioned. Forum link sent me to a website placeholder..? Do you have a link or a cut/paste you
can sharing?
|
|
|
Mush
National Hazard
Posts: 633
Registered: 27-12-2008
Member Is Offline
Mood: No Mood
|
|
1, The Alkylation of Phenols with Dialkyl Oxalate. III. n-Propylation, n-Butylation and Isoamylation
Yoshiaki Sakakibara
Nippon kagaku zassi Vol. 82(1961) No. 3
2,
The Alkylation of Phenols by Means of Oxalate. II. The Methylation and the Ethylation of Phenols
Yoshiaki SAKAKIBARA
Nippon kagaku zassi Vol. 81(1960) No. 3
Science reports of the Yokohama National University. Section 1, Mathematics, physics, chemistry 19 ,p32-47,1972
3, Allylation of Phenols (with diallyl oxalate)
http://kamome.lib.ynu.ac.jp/dspace/simple-search?query=Yoshi...
4, N-Methylation and N-Ethylation of Aniline
http://kamome.lib.ynu.ac.jp/dspace/simple-search?query=Yoshi...
5, n-Octylation of Phenols (with di-n-octyl-oxalate)
http://kamome.lib.ynu.ac.jp/dspace/simple-search?query=Yoshi...
Attachment: 2, Nippon kagaku zassi Vol. 81(1960) No. 3 81_495.pdf (1.3MB)
This file has been downloaded 1024 times
Attachment: 1, Nippon kagaku zassi Vol. 82(1961) No. 3 82_403.pdf (835kB)
This file has been downloaded 948 times
[Edited on 19-4-2015 by Mush]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Nein, nicht understandu, neither word. Can anyone at least quote the conditions and yields for the methylation via dimethyl oxalate? Because google
translate doesn't help to undarstand what paper says: "Death in the previous report yu "as a firewood seems alkylating agent
Oxalic acid dimethicone". The problem is a bad characters recognition in the original document.
At hive people mentioned that the method gives really low yields.
Btw, does the next article at the end of 2, Nippon kagaku zassi Vol. 81(1960) No. 3 81_495.pdf tell about amination of alcohol via imide?
For some reason there's not much said about dimethyl carbonate, while it is a relatively ease to synthesize via ethylene carbonate and further
transesterification with methanol via basic catalyst like NaOH or K3PO4.
The stumbling-stone of the methylation with carboxylic acid ester is that phenols prefer to give acid-phenol ester rather than the alkyl-phenol one.
Same difficulty exist while methylating with dimethylcarbonate, and the solution is heat to at least 150°C driving away CO2. As you may understand,
you can't get rid of the oxalate, so the same trick doesn't work for dimethyloxalate.
UPD: oops, looks like the OP article (dimethyloxalate.pdf) has a translated description of substrates and yields. So the yield for phenol
o-methylation is almost 80%.
Here's an article in english describing the mechanism and a picture from Houben-Weyl which is actually the same as in the paper, but is easier to
read.
As you may notice, the actual mechanism is not a transesterification, and you can't use any other solvent except formic secondary amides (which is DMF
in most cases).
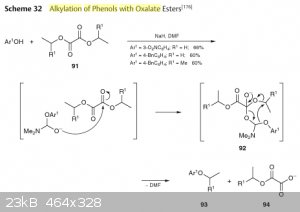
Attachment: Phenols alkylation with alkyl oxalate (271kB)
This file has been downloaded 902 times
[Edited on 19-4-2015 by byko3y]
|
|
|
Mush
National Hazard
Posts: 633
Registered: 27-12-2008
Member Is Offline
Mood: No Mood
|
|
Qing-hua. C.; Ju-xian. L.; Lan-gui. C.; Xue-wen. 2. Acta Chimica Sinica 1981,39,263
THE ALKYLATION OF PHENOLIC HYDROXYL GROUP IN BERBAMINE
CHEN QING-HUA, LU JU-XIAN, CHEN LAN-GUI, ZHAO XUE-WEN
Institute of Hygiene, Chinese Academy of Medical Sciences, Beijing
Abstract A method concerning the selective alkylation of phenolic group in berbamine by means of dialkyl oxalate was reported. We succeeded in
preparing methyl, ethyl and iso-propyl ethers of berbamine without N-alkylation by treating sodium berbaminate with the corresponding dialkyl
oxalates.
Sorry, this paper is written in Chinese. 
Thanks for that scheme! Here is the article what the book referes to.
(176) Anomalous ether formation in attempts to transesterify oxalate esters with phenoxides
Edward E. Smissman, Michael D. Corbett, Samir. El-Antably, Kathryn C. Kroboth
JOC, 1972, pp 3944–3945
DOI: 10.1021/jo00797a040
Attachment: THE ALKYLATION OF PHENOLIC HYDROXYL GROUP IN BERBAMINE.pdf (2.5MB)
This file has been downloaded 889 times
Attachment: Anomalous ether formation in attempts to transesterify o.pdf (293kB)
This file has been downloaded 865 times
[Edited on 19-4-2015 by Mush]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I dunno how to tell ya but... it's the same paper as I posted. Look at the file size.
Trying to translate the first procedure from the chinese paper:
"1. Dimethyl oxalate ester method: 50 ml of anhydrous methanol in a dry three-necked flask, 0.46 g (0.02 mol) of sodium metal mole, 12.2 g (0,02 mol)
small polyamine was added after the completion of action , heated and stirred to dissolve.
The methanol was evaporated, 60 ml of toluene, 10 ml of toluene was distilled off, coolish, ester was added 4.7 g of oxalic acid dimethyl [11], (0.04
mol), heated at reflux for 3 hours identified by thin layer chromatography, about 75% the amine was converted to ether small bi. Was extracted with 2%
hydrochloric acid, then with 2% potassium hydroxide aqueous solution was basified to ph 10, deposited precipitate was filtered, the precipitate washed
with water until neutral, 60C and dried to give 9.4 g of a white powder , yield 75.5% acetone was purified by recrystallization or column, you can get
4.7 grams of fine granular colorless crystals, mp 180 ~ 182 ° С, the yield of 37%;. Specific rotation [a] 15D + 150 ° (c. 0.89, chloroform); Rf
value Rf0.83 ([3] reported that the melting point of 182 ~ 183 ° С (acetone); specific rotation [a] 20D + 151 ° (c 0.85, chloroform))"
So they've got 75% crude yield using NaOMe and dimethyl oxalate. I can't see the DMF there in fact, nowhere in the article.
UPD: Forgot to link a nice article about dimethyl oxalate preparation:
"Protic Acid Immobilized on Solid Support as an Extremely Efficient Recyclable Catalyst System for a Direct and Atom Economical Esterification of
Carboxylic Acids with Alcohols"
A. K. Chakraborti, B. Singh, S. V. Chankeshwara, A. R. Patel, J. Org. Chem., 2009, 74, 5967-5974.
DOI: 10.1021/jo900614s
http://www.organic-chemistry.org/abstracts/lit2/638.shtm
At the official page you can freely download a supporting information file containing the procedure ( this one http://pubs.acs.org/doi/suppl/10.1021/jo900614s/suppl_file/j... )
Briefly: HClO4 on silica gel powder (obtained by adding HClO4 to a silica gel in Et2O, evaporating the ether and then heating for 3 days at 100 C in
vacuum) can be used to catalyze the esterification of a hell lot of alcohols and acids. To perform the reaction you need to just mix everything (acid,
alcohol and catalyst) and stir few hours until it's done.
The orgsyn procedure gives lower yield and requires some nasty method to separate the ester. Though I don't know if the HClO4 catalyst is applicable
to dimethyl oxalate.
[Edited on 19-4-2015 by byko3y]
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
I wonder if you couldn't find some sort of drying agent that would let you esterify methanol and toluenesulfonic acid. It's possible to get TsOMe by
reacting TsOH with a methyl orthoester which can be produced from methanol and acetonitrile. Maybe you could do the same thing with methyl carbamate:
MeOCONH2 + 2TsOH >> MeOTs + CO2 + NH4OTs
For methyl orthoacetate see the attachment on preparing orthoesters from nitriles and alcohols. Acetonitrile, of course.
Then TsOH + Et(OMe)3 >> TsOMe + MeOH + AcOMe:
https://www.erowid.org/archive/rhodium/chemistry/sulfonic.es... (I am being a little optimistic that orthoacetate will behave like
orthoformate...)
Attachment: php7AVSWT (335kB)
This file has been downloaded 973 times
[Edited on 20-4-2015 by clearly_not_atara]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
What will hydrolyze the methyl carbamate? At the left side you have an amide, at the right side there are acid and ammonia.
Methyl tosylate is OTC if you can get toluenesulfonic acid and either acetonitrile or sodium methoxide + chloroform, which looks more OTC to me,
though longer to perform.
And yes, orthoacetate is almost the same as orthoformate. Orthoformate is employed commonly because it can be cheaply made industrialy using mentioned
sodium alkoxide + chloroform, while acetonitrile by itself costs a lot.
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Quote: Originally posted by byko3y  | | What will hydrolyze the methyl carbamate? At the left side you have an amide, at the right side there are acid and ammonia. |
Henry Louis le Châtelier, of course. MsOH transesterifies to give methyl mesylate and carbamic acid HO2CNH2, although the equilibrium is in favor of
methyl carbamate, and carbamic acid decomposes to CO2 and NH3. We have to hope MsOMe doesn't react with NH4OMs, which normally only happens under
basic conditions / high temps. Water shouldn't be necessary.
Also, methanesulfonic acid or ethanesulfonic acid makes more sense because those are liquids. Ethanesulfonic acid can be had from EtBr + Na2SO3
(Strecker sulfite alkylation).
[Edited on 20-4-2015 by clearly_not_atara]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
No, it would not. I see no reason for tosyl to attack the ester oxygen, even if it's converted into oxonium. Water can easily hydrolize this but not
the tosyl. Remember that tosyl is a good leaving group, so you will have a hard time trying to attach it to anything.
Although the tosyl could have attacked the carbonic acid carbon, but again the tosyl s a good leaving group.
Toluenesulfonic, methane- and ethanesulfonic acids have pretty similar functional properties.
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
You're forgetting that it's an equilibrium.
MsOH + MeOCONH2 <<<< > MsOMe + HOCONH2 (maybe the left is favored by 10^5?)
HOCONH2 >>>> CO2 + NH2 (incalculably huge; MsOH's affinity for ammonia makes this irreversible)
Everything is assisted by using an excess of MsOH. Now, maybe carbamic acid is stabilized somehow, or maybe MsOMe reacts with NH2 regardless, but it
conceivable.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Carbamic acid has pka close to acetic acid. So basicall it's MsOH + MeOAc <-> MsOMe + HOAc, with equilibrium strongly to the left, because MsOMe
is highly electron deficient and HOAc is electron sufficient. I want to emphasize that the mixture on the right side is not slightly acidic, but
strongly acidic because the MsOMe is a lewis acid in fact with the oxygen positively charged.
Decomposition product of carbamates should be close to urea's ones (cyanuric acid, ammelide and ammeline http://www.sciencedirect.com/science/article/pii/S0040603104... )
I like the idea of driving the equilibrium by evaporating the reactants, but I don't think it's possible here.
Actually, toluenesulfonic acid is a good transesterification catalyst for converting dimethyl carbonate into diphenylcarbonate ( http://www.sciencedirect.com/science/article/pii/S1566736710... ), though in this reaction amount of methyl tosylate is close to zero.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Dimethyl oxalate decarbonylation to dimethyl carbonate
US4544507 - alkoxides
US5973184 - alkali carbonates
(also CN 1113852)

[Edited on 1-5-2015 by byko3y]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I've tried esterification of oxalic acid via HClO4-SiO2 as the article http://pubs.acs.org/doi/abs/10.1021/jo900614s says, and I'd say it's ludicrous. My first suspicion aroused after reading about esterification of
methanol at 60-80°C in an open reactor.
And truly, the goddamn methanol just flew away. 1-5% yield of dimethyl oxalate is what I've got. I used oxalic acid dyhidrate, but what is the point
of the catalyst which doesn't help to get rid of water?
I'm starting to feel like I can ignore every article written by indian "researchers".
There's a nice review of esterification reactions
http://www.hindawi.com/journals/isrn/2012/142857/
and basing on it I can say that a sulfuric acid is a best catalyst you can pick for fisher esterification of oxalic acid. In fact, you can use a plain
oxalic acid as a catalyst, though you will need a lot of time to reach equilibrium. Actually, you can use almost any lewis or bronster acid, like HCl,
NaHSO4, BF3, SnCl2, etc.
But the main problem with the dimethyl oxalate is that it's the only case when you can't dehydrate the reaction mixture easily. You can use
carbodiimides for that, or thionyl chloride and product of its reaction with silica gel (so-called silica chloride and methoxyl silica gel), or maybe
polyphosphoric acid, but the authors of orgsyn article use sulfuric acid for dehydration: 0.64 mole of H2SO4 per 1 mole of oxalic acid and 2.5 moles of methanol, this is clearly not
just a catalyst, but a dehydration agent. And if you use too much of sulfuric acid - your mixture will become viscous and it becomes harder to
separate the product.
This rises some questions:
- would it be easier to use monomethyl sulfate directly instead of using oxalate as alkyl carrier?
- in case of phosphoric acid as a dehydration agent (polyphosphoric/P2O5), we have n-methyl-phosphates as intermediates ( e.g. US4133838 ); maybe it's
better to avoid the dimethyloxalate and go strait to the trimethyl phosphate?
As far as I can see, dimethyloxalate is viable in case you produce it from something like methanol and CO on palladium catalyst, which is clearly not OTC. And within OTC constraints every route to dimethyloxalate lies through some
methylation agent.
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
The examples given in the paper are about equimolar mixtures of methanol and benzoic acid. If I got it right, you have used 2 parts water, 1 part
oxalic acid, and 2 parts methanol. So you didn't do what the paper says but something different.
| Quote: | | My first suspicion aroused after reading about esterification of methanol at 60-80°C in an open reactor.And truly, the goddamn methanol just flew
away. 1-5% yield of dimethyl oxalate is what I've got. |
By an open reactor you mean non pressurized with reflux?
Maybe the boiling point of methanol is elevated due to the highly concentrated mixture with benzoic acid, so it doesn't fly away. If a diacid is used
as the substrate the molality of the methanol mixture is only half compared to a monoacid.
| Quote: | | I used oxalic acid dyhidrate, but what is the point of the catalyst which doesn't help to get rid of water? |
Maybe one equivalent of water can get adsorbed by the silica effectively, which does not mean more water could get absorbed equally well.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Alice, I did 10 g of SiO2 + 1.75g of HClO4 (17.5 mmol, almost 2% by methanol) catalyst for 55 g of oxalic acid dyhydrate (0.45 mole) and 32 g of
methanol (1.0 mole). After reaction, an ether extract gave me something that can be called traces of ester.
I did put a simple reflux condenser on it, and obviously it did not pressurize the vessel, and probably some amount of methanol was left in the
reaction mixture, but it's not enough to make any decent yield.
Methanol-benzoic acid mixture is zeotropic, as well as benzoic acid-water. You can keep the mixture on reflux without methanol flying away, ut you
won't be able to reach 80°C because it starts bumping as crazy, and again your methanol will be out of reaction in the reflux condenser all the time.
If you use a strong acid, like nitrobenzoic or oxalic, then the equilibrium is shifted to acid+alcohol, while for ethanol and acetic acid equilibrium
constant is like [ester]*[water] / [alcohol]*[acid] = 4. And this is why dialkyl oxalates are used for alkylation - they can easily give away the
alkyl group.
Silica gel absorbs 0.2g/g (0.1 mole of water for my case) at room temperature, which is far even from absorbing water of esterification water.
It's okay, I was performing bulshit researches in my college and writing reports about them. It's just sad that indian researches are forced to
publish their articles in magazines like tetrahedron or synlett.
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Thank you for mentioning the equilibrium issue. But this means that an esterification reaction where water is not removed sufficiently (like the one
you tried here) is a bad idea with strong acids like oxalic acid. The authors didn't show the reaction for oxalic acid anyway.
Comparing the amount of catalyst you used with the amount the authors used shows some difference:
Paper example:
50 mmol n-octanol -> 1 g catalyst ("1%")
For 1 mol n-octanol -> 20 g catalyst
Your example:
1 mol methanol -> 10 g SiO2 + 1.75 g HClO4
So you have used another catalyst composition. Let's see what is the difference in catalyst preparation:
Paper:
23.75 g SiO2 + HClO4 1.25 g (12.5 mmol)
You:
10 g SiO2 + 1.75 g HClO4 (17.5 mmol)
The factor of HCLO4 content is 3.3 between yours and the catalyst described in literature.
If you like to perform the reaction like stated in literature with "2 %" you would have to use 40 g of catalyst for 1 mol methanol.
I didn't search for a value of SiO2 water adsorption, so thanks for delivering one. This speaks against water removal as a major driving force but it
might still contribute to some degree especially if the equilibrium is already on the product side.
I think your experiment is not suitable to make any judgements about this publication. Unless an experiment is performed according to one of the
literature examples I remain undecided. 
[Edited on 12-5-2015 by Alice]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Basicall, esterification of methanol with benzoic acid will work for any acidic catalyst. The truth is: you don't need HClO4-SiO2 catalyst to get
decent yields, and the procedure for esterification of those particular reagents is wrong. You need to heat the reaction mixture to methanol reflux,
and this is the highest temperature you can reach.
I want to emphasize that the stochiometric amount of methanol is 0.7 mmol, so I was very close to the article conditions in that regard.
http://pubs.rsc.org/en/Content/ArticleLanding/2003/CC/b30417... - The original "research" on the catalyst. First, the authors (which are all the
same people as with other article) used aqueous HClO4 and it worked well (for Ac2O acylation, lol). There's neither rationalization nor experimental
data on stochiometric of the HClO4-SiO2 catalyst, they just used the only perchloric acid loading all over their works. Basically, you can add aqueous
HClO4 and the reaction should work too, though in the esterification article the authors never compared free and supported acids.
First and foremost difference of my conditions and article conditions - I've used a hydrated reagent. But what is the point of the catalyst, if a
catalytic amount of sulfuric acid can give you the same results?
|
|
|
| Pages:
1
2
3
..
5 |









