| Pages:
1
2 |
DerAlte
National Hazard

Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Electrolytic Production of Sulphuric Acid + some questions.
Sulphuric acid is a uniquely useful acid in both organic and inorganic chemistry due to its high boiling point, ionic strength, and dehydrating
properties. For the amateur chemist it is becoming increasingly difficult to obtain, even in impure forms. The Drain cleaner type, of questionable
quality, is being phased out. Once one could go to any service garage and buy a gallon of pretty pure acid of density 1.3 (40%) for a few dollars.
A catalog for MCB for c 1975 quotes the price of reagent grade, 95-98%, al impurities at < 2ppm, as 2lb (0,9kg) for $5.90 and fuming (30% SO3)
oleum at $11.38/lb. Compare today’s prices! (But allow for inflation).
The subject of H2SO4 production has received much attention in these pages (UTSE). I recently did a few electrolyses to produce dilute acid. The aim
was to produce acid of acceptable purity which could be concentrated if required to the 95% level.
These electrolyses were carried out in cells using a hole-less clay pot as diaphragm and an iron cathode. The ideal anode would be Pt – forget it! I
tried carbon, but the erosion and /or oxidation rate was worse, if possible, than with the chlorate process so familiar to you all. For the first try
with carbon anodes I used Na2SO4 at current density 250 ma / cm^2. A small amount of acid was produced but the experiment halted due to massive anode
erosion.
I then decided to use lead for the anode. Lead oxidizes to dioxide, but the dioxide coats the lead and is a good conductor (think of the +ve plate of
a lead acid accumulator). The results were reasonably satisfactory, resulting in crystal clear acid of about 20% strength.
The Pb immersed area was about 30 cm^2, and the average current density ~ 120 ma/cm^2. The Pb anode was a thin strip (0.8mm thick) and if anyone
tries it, I strongly recommend a greater thickness – the strip must be handled very carefully to avoid the PbO2 coat being disturbed.
The outer 1 liter container was filled with a saturated solution of commercial NaHSO4 containing 8% Na2SO4 (No problem there); initially the inner
was filled with boiled city water (main contaminant Ca++, about 40 ppm; all others <= 1ppm except Na+ @ 10ppm). The clay pot was saturated with
water and allowed to percolate for 2 days before use.
The initial current was very low, ~20 ma for 16 volts across the cell, the drop occurring mainly across the anode compartment. This rose slowly, as
expected. The lead gradually oxidized to a brown black color before significant oxygen was evolved; hydrogen evolution was ab initio. The current was
controlled with series resistance, including a tungsten lamp, and switched voltages from the PSU.
I cheated by introducing a small amount of H2SO4 (actually from the carbon run) to enhance the conductivity of the anode compartment, which took
nearly an hour and a half to get to 100 ma. The current then increased exponentially and was controlled to about 2 Amps initially, and later allowed
to rise to 3.5 amps.
Some dioxide spalled off the anode during the run and the lead became fragile and eroded. 9 gm of PbO2 was collected finally off the anode and in the
liquid. At least PbO2 is very heavy and can be removed by decanting, unlike the filthy carbon.
I have not yet tested the acid for purity.
My major question concerns the coulomb efficiency. If all the conduction was due to HSO4- ions this was only 30%. Why so low? Any ideas?
Can anyone suggest a better anode than lead? What about Ni?
Regards, Der Alte
|
|
|
chloric1
International Hazard
Posts: 1141
Registered: 8-10-2003
Location: GroupVII of the periodic table
Member Is Offline
Mood: Stoichiometrically Balanced
|
|
well lead would be my first choice based on availability and cost. Try melting lead into a massive rod form. Should last longer. Another anode of
consideration would be baked spinel coated titanium. Nickel would probably work intially but there would be nickel build up I would think once you
gained acid concentration.
About the porous memebrane method, your acid might never lose all of its sodium since 100 % seperation never occurs. To get thorough separation, you
need a nafion membrane (expensive)!
Another option is to obtain a strongly acid ion exchange resin and convert it to the hydrogen form then run ammonium sulfate through it. Ammonium
sulfate is very water soluble unlike its potassium and sodium counterparts.
Fellow molecular manipulator
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
I have some lead 1,7 cms square of unknown purity and intend to use it on my next run. The first was proof of concept.
The migration of a few Na+ ions is possible but one thing I did not mention is that osmotic pressure tends to keep the water free of it. At first I
was surprised at the speed with which the water in the anode compartment was consumed - then it dawned on me!
I wonder if a moderator could put this thread in its rightful place - in Technochemistry? I wasn't watching when I put it in this section.
Regards, Der Alte
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
I evaporated to dryness around 10cc of the acid produced by the carbon electrode, after boiling to about 40% H2SO4. . (Don't try this indoors - even
this small amount produces copious fumes of SO3/H2SO4). White solid residue, assumedly Na2SO4 or pyrosulphate, amounted to less than or around 0.2% Na
by weight of H2SO4.
In the next few days I will try the lead bar as anode (about 50 cm^2 area). Need to prepare a better power supply also...
Der Alte
|
|
|
Nightscape
Harmless
Posts: 1
Registered: 12-9-2008
Member Is Offline
Mood: No Mood
|
|
Would this work with Ammonium Sulfate or am I missing something?
I am currently working on producing Sulfuric acid by thermal decomposition of Ammonium Sulfate dissolved in H2SO4.
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
It would. Ammonium doesn't have anything to do with the anode side. You do get ammonia at the cathode, however, which will have to be well
contained, preferrably sealed (it can reach a high strength if kept cold, but has a high vapor pressure too!). Of course you have hydrogen sparging
it out, so you'd be best off running it through a scrubber I guess.
Edit: ammonium *bi*sulfate disproportionates to H2SO4 and (NH4)2SO4, which of course doesn't have a significant vapor pressure.
Tim
[Edited on 9-12-2008 by 12AX7]
|
|
|
OMG
Harmless
Posts: 15
Registered: 26-7-2008
Location: BC, Canada
Member Is Offline
Mood: No Mood
|
|
I read a patent once that regenerated sulfuric acid using a two cell setup. The anode was lead, the anolyte was ammonium sulfate, the cathode was
iron, the catholyte was spent acid (iron sulfate I think). It did use a nafion membrane, but clay might work. During the process, the ammonium ion
migrated to the catholyte and the iron plated out, and left just the sulfuric in the anolyte.
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Sulphuric Acid, contd.
I have now done a series of runs resulting in the equivalent of 520 g of 100% acid (as dilute acid, 27% and 40% batches). As noted previously, the
coulomb efficiency seems low, and averaged 0.42 g/Ah, i.e. about 1240 Ah were consumed to produce this. This is around 8 days at the current of 6A!
11KWH est. consumed
and patience required.
I have discovered (I think) the reason why the efficiency is this low. The anode cell contains mainly H2SO4 which ionizes as
H2SO4 --> H+ + HSO4- (Ka1 ~ 10^3);
the second ionization is relatively weak:
HSO4- --> H+ + SO4- - (Ka2 ~ 1.2x10^-2)
The ion current can be considered as being carried by H+ (toward cathode) and HSO4- (to anode) in the anode cell, regardless of the ions in the
cathode cell, provided either HSO4- or SO4- - are present, and no other negative ions which might migrate to the anode. In the anode cell, then, the
relative mobility (directly related to conductivity) of the H+ ions is about 350 and HSO4- about 50, (units 10^-4 m^2 mho/mol).(CRC). For every HSO4-
ion passing through the barrier, 7 H+ ions go the other way, per 8 electrons delivered to the anode. This means that 7 HSO4- ions deliver their
electron from the anode solution for every one that comes from the cathode area. These ions produce oxygen (or an oxide) at the anode by some such
process as:
HSO4- --> ‘HSO4’ + e-
2 ‘HSO4’ + H2O --> 2H2SO4 +’O’ (needs 2e-)
2 ‘O’ --> O2 (needs 4e-);
And, of course, the H2SO4 re-ionizes to H+ and HSO4-
So, if you tot up charges since there is not charge accumulation, you find that of 8 coulombs, only 1 carries our desired HSO4- ion from the cathode
compartment to enrich the anolyte. Faraday’s Law remains intact! In effect, we are mainly electrolyzing water.
The expected efficiency for this process is one mol per 8F, or in terms of Ah, 3600/96500/8 = 4.66.10^-3 mol/Ah. For HSO4-, 1 mol=97 g, so we expect
an increase of 0.452 g/Ah. This is close to what I got.
A few tips if you attempt this:
(1) Precaution; Do not do in any enclosed space ; ensure that the gases involved are rapidly dissipated, e.g. by a fan. The coaxial
arrangement of the cells could mix H2 and O2 into a dangerously explosive combination.
(2) Cover the anode cell with a lid to prevent spray from the cathode cell contaminating the anolyte. Arrange a vent (plus a pipe, if possible) to
allow O2 to exit, away from H2 from the cathode.
(3) Since water is lost from both cells by the electrolysis, make up from time to time. But it helps to allow the anolyte level to decrease at the end
of a run to concentrate the acid.
(4) Use a constant current source if available. The resistance of the cell decreases rapidly with time at first, and also drops as the temperature
rises. The resistance of my arrangement was about 0.37 -0.5 ohm; OC cell EMF about 2,2 V for bisulphate and 2.05 V for sulphate.
(5) Do not attempt to concentrate to much more than 20% acid in the cell. Efficiency drops off as concentration increases (haven’t worked out why,
experimental fact). For >20% see next post.
(6) Lead is consumed. I recovered 20g of PbO2 from the above runs. A fat round anode would probably be best; mine was square and tended to loose
coating at the corners.
(7) Make the cathode cell considerably larger than the anode. Do not space the cathode close to the anode or the catholyte could became depleted in
HSO4- ions locally.
(8) NaHSO4 solubility is about 50g/100g aq @ 0C, around 75 @ RT and ~100 at 100C. Keep the catholyte concentrated. It effectively gets converted to
Na2SO4 as it is consumed. NaSO4 is not as soluble and may precipitate out. If run to depletion, NaOH results. The change can easily be detected by an
indicator. Na2SO4 will give equally good results.
(9) Use an iron cathode. I tried stainless steel, low Cr, but it seems to react more with the acid sulphate, gives rapid evidence of Cr++ ions (blue).
Iron dissolves too, of course. Once the catholyte goes alkaline, Fe2(OH)3 precipitates (Stainless steel then changes the Cr++ to Cr+++, I think. Color
changes to green). Never leave the cathode in the liquid, or leave the anode compartment in the catholyte, with the current off. Contamination will
result by diffusion. Don’t leave the anode in its cell either – sulphation will result.
Regards, Der Alte
|
|
|
chloric1
International Hazard
Posts: 1141
Registered: 8-10-2003
Location: GroupVII of the periodic table
Member Is Offline
Mood: Stoichiometrically Balanced
|
|
Great work DeAlt! Right noe, it would be a project of only academic concern since I am still able to obtain automotive battery electrolyte at
reasonable cost. But, having a workable plan as back up is getting more practical as global governance tighens its death grip on us all.
A few point and concerns if you will:
Since the method only produces 20% concentration with any respectable efficiency, boiling off to concentrate the acid is going to be a major endeavor.
With battery electrolyte of 35 to 40% initial concentration, it is necessary to boil down to ONE FOURTH original volume just to obtain acid of
specific gravity of 1.84! So if you want respectable amounts of 98%, then you will need MASSIVE amounts of the 20%. So if you want respectable amounts of 98%, then you will need MASSIVE amounts of the 20%.
Also, there is a potential for significant acid losses due to varying combinations of water and H2SO4 that boil at different temperatures. So, it
might be pertanent to distill the volatiles into a receiver as apposed to just "boiling off".
There seems to be a variation to the cells resistance during startup and full run. Maybe just hooking up a high powered Variac transformer with a
bridge rectifier circuit to like 3 or 4 cells in series might help this. Start with 16- 20 V per cell then as the current climbs, turn down the
voltage. A 1000W dimmer might even be handy here.
Now questions:
DeAlt-do you have data on current density at your cathode and anode? What temperatures did you run at?
Secondly- I am out of time, could someone find some data on the boiling points of the various H2SO4/water combinations?
Thanks in advance
Fellow molecular manipulator
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Chloric wrote:
| Quote: | | Since the method only produces 20% concentration with any respectable efficiency, boiling off to concentrate the acid is going to be a major endeavor
|
Absolutely! I was going to write a follow-on post on concentration and will do so when time permits (too busy with this bloody stock market and family
crises).
| Quote: | | DeAlt-do you have data on current density at your cathode and anode? What temperatures did you run at? |
100-200 ma/cm^2, temp around 40C depending on current. The anode cell gets hotter than that.
| Quote: | | ...as global governance tighens its death grip on us all.... |
Sulphuric acid is on the DEA diversion list II and also has been noted by the UN. Quantities involved are probably large and concentration high, I
don't have the details.
....some data on the boiling points of the various H2SO4/water combinations
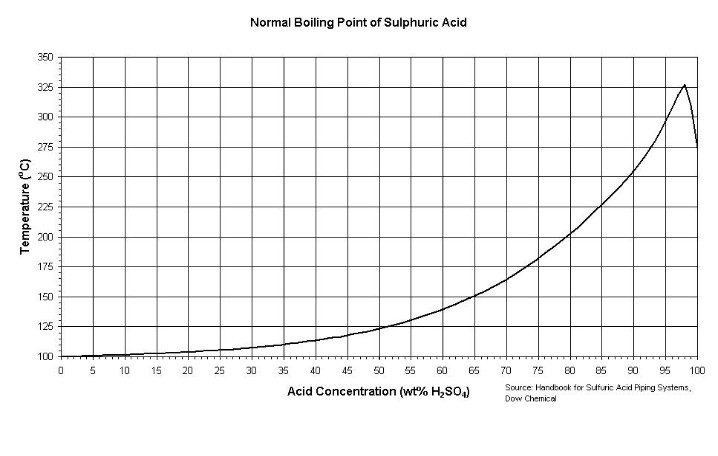
I have a whole lot more data on H2SO4 that I gathered in this investigation - but that will have to be later...
Regards, Der Alte
|
|
|
Taoiseach
Hazard to Others
Posts: 241
Registered: 16-3-2008
Member Is Offline
Mood: No Mood
|
|
Awesome work again, DerAlte! Does this work with FeSO4 or Na2SO4 too? The latter is tempting because plaster of Paris could be used as a source of
SO4(2-): CaSO4 + Na2CO3 => Na2SO4 + CaCO3 
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Sure you can! FeSO4 (and Na2SO4) will work. FeSO4 will gradually convert to Fe2(SO4)3 as electrolysis proceeds so the catholyte will become yellow to
brown. Fe will then then deposit on your iron cathode. Any soluble sulphate/hydrogen sulphate will work. Copper will deposit metallic copper. However,
some like magnesium or calcium will produce insoluble products at the cathode and perhaps in the porous barrier. Calcium sulphate is too poorly
soluble , for instance; magnesium will precipitate a mess of hydroxide. Best to keep to the alkali metal sulphate/bisulphates.
Der Alte
|
|
|
texaspete
Harmless
Posts: 28
Registered: 26-7-2006
Member Is Offline
Mood: No Mood
|
|
I did this once sort of accidentally with Cu2SO4. Cu is plated out on the cathode, leaving sulfuric acid. I left some of this copper sulfate/copper
metal out to dry, and the resulting liquid reacted with sodium bicarb.
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
I've attempted to plate copper out of an acidic copper chloride solution, which worked more or less (leading to rather nice crystals rather than a
clean plated surface), and producing chlorine instead of oxygen at the anode.
Tim
|
|
|
chloric1
International Hazard
Posts: 1141
Registered: 8-10-2003
Location: GroupVII of the periodic table
Member Is Offline
Mood: Stoichiometrically Balanced
|
|
| Quote: | Originally posted by texaspete
I did this once sort of accidentally with Cu2SO4. Cu is plated out on the cathode, leaving sulfuric acid. I left some of this copper sulfate/copper
metal out to dry, and the resulting liquid reacted with sodium bicarb. |
Keep in mind that sodium bicarbonate reacts with copper sulfate anyways to produce basic copper carbonate, sodium sulfate and carbon dioxide gas. If
you use hot concentrated copper sulfate solution and you let the resulting products cool to near freezing you will see nice long needleshaped crystals
of sodium sulfate decahydrate.
I was pondering if the same setup could be used with monobasic sodium phosphate to produce phosphoric acid. I would think that the lead anode would
still form a PbO2 coating and the phosphoric acid is dilute so it should not attack glass or ceramics. Besides to attack these materials,
concentrated phosphoric acid needs to be heated to 200 or 300 Celsius anyways.
Fellow molecular manipulator
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Sulphuric Acid, contd.: Concentration, etc
As Chloric pointed out, a lot of effort is needed to concentrate weak sulphuric acid. The acid is somewhat unusual in that anything below around 75%
is called dilute. This is about 13M. Pure acid is 18.6M.
For general reagent purposes a 10% acid (1M) often suffices. For such things as producing nitric acid, HCl, SO2, etc, around 70-95% is desirable. And
for organic chemistry, in such operations as chemical dehydration or as a catalyst, the stronger the better. As a dessicant it is second to none
(almost) but must be >80% for effectiveness (partial pressure of H2O is then about 10pa (n/m^2 in MKS) or 0.08mm Hg.@ 20C).
To estimate the strength nothing beats titration, but a hydrometer is very useful. If you know the strength of a sample, you can estimate what you
have after concentration by comparing volumes. Sulphuric acid is essentially non-volatile, like phosphoric acid. Here are a few figures I have
calculated for the relative volumes of solutions containing the same amount of acid:
2%, 90.9; 5%, 35.7; 10%, 17.2; 20%, 8.03; 50%, 2.62; 70%, 1.63; 80%, 1.33; 95%, 1.05; 100%, 1.00
You cannot concentrate beyond about 97% by heating. At that temp. it forms a sort of constant BP mixture which is actually a decomposition product
into H2O and SO3. This will recombine at a lower temp, so in a sense, H2SO4 can be ‘distilled’ to get about 97.5%, but the pure acid cannot be
gained by heat alone. You have to add SO3 to partially hydrated acid to get 100% or oleum (see thread on this; UTSE.)
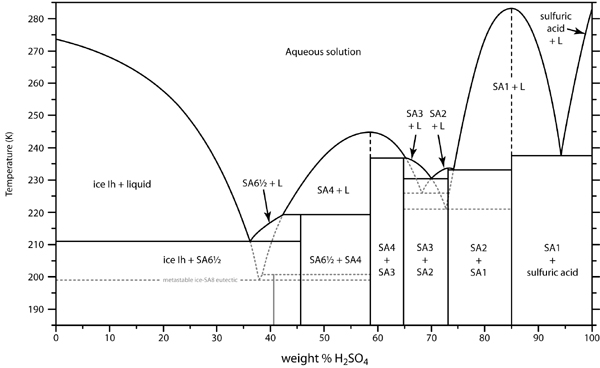
H2SO4 forms distinct hydrates. You can actually concentrate weak acid (<37%) by freezing. I have tried it. It is not a practical method because the
ice forms as a gelatinous slurry, almost impossible to separate. I saw a patent for doing this, by centrifugation. Also, the monohydrate H2SO4.H2O
(84.4% by weight), freezes out at 8.6C, a tetrahydrate at about -33C. See the freezing curve:
Heating in a microwave oven works well, up to the point where the vapor pressure of H2SO4 becomes high enough to cause corrosion. Partial pressure of
the acid (not the water component) at the boiling temperatures is approximately as follows;
80% BP ~205C PP of acid ~ 0.3mm Hg.
74% BP ~180C PP of acid ~ 6*10^-2 mm Hg.
67% BP ~160C PP of acid ~ 4*10^-3 mm Hg.
62% BP ~140C PP of acid ~ 2*10^- 4 mm Hg.
So anything below 70% should not produce noticeable fumes (only steam).
One problem with the MW oven is the very high dielectric const of H2SO4 (~100) plus the very high conductivity of conc. acid. This tends to put a high
stress on the magnetron (due to mismatch) which is likely to run hot and may trip the thermal breaker which some have – or blow the magnetron/and or
PS in a cheapie.
Traditional boiling in an open vessel works well. I have had little bumping until you get to around 70%. Use a bit of unglazed porcelain or even
silica sand to aid stable boiling. And be very careful of the hot acid (over 300C) when it gets near maximum concentration of 95+%, where the vapor
pressure is over 75 mm Hg, producing copious noxious acid fumes. I saw a picture once of what hot conc. acid does to a hand – not pretty.
A convection oven works slowly and is less efficient, but the movement of vapor off the acid by the fan helps evaporation. There is the same problem
of possible corrosion, so 70% odd is about the limit.
Finally, the amount of heat required to evaporate off water is rather high. I calculate, crudely, that to evaporate 1g acid in a 20% to a 95% solution
takes about 12KJ. This includes the heat required to bring to temp., enthalpy of evaporation of water and the enthalpy of dilution which has to be
returned on concentration. Thus, although the electrolysis is not very efficient beyond 20%, the price on concentrating is such that one might as well
go to 30% or even higher and concentrate as far as possible that way, by electrolyzing off the water! Pity one can’t use the H2 and O2 in some way,
it seems very wasteful to just let them dissipate.
A lot of work for half a liter of acid! Enough said,
Regards, Der Alte
|
|
|
chloric1
International Hazard
Posts: 1141
Registered: 8-10-2003
Location: GroupVII of the periodic table
Member Is Offline
Mood: Stoichiometrically Balanced
|
|
With both cathode and anode made of lead, in an undivided cell, should concentrate to 40% or more with little trouble. So run your divided cell to
separate your 20% acid then transfer anolyte to a beaker or pyrex dish and electrolyze between the lead electrodes. This will be quicker than
continuing the intitial run I would think.
Fellow molecular manipulator
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Correction and Further ideas
Der Alte wrote, above:
| Quote: | | In the anode cell, then, the relative mobility (directly related to conductivity) of the H+ ions is about 350 and HSO4- about 50, (units 10^-4 m^2
mho/mol).(CRC). For every HSO4- ion passing through the barrier, 7 H+ ions go the other way, per 8 electrons delivered to the anode. This means that
7 HSO4- ions deliver their electron from the anode solution for every one that comes from the cathode area. |
On re-reading, this conclusion is fallacious and the reasoning distinctly flaky! Mobility has nothing to do with the ionic transfer across the barrier
diaphragm. It does affect, transiently, the concentration near the electrodes, but the electrolyte is constantly stirred by gas emission at them,
creating bubbles and convection. Although local concentrations may be affected, the bulk solutions, on average, are not.
My revised analysis for the cell as previously described above is:-
1. At the anode compartment (hopefully) all we have is dilute sulphuric acid, ionizing mainly as follows H2SO4 --> H+ + HSO4-, Ka1 ~ 1e3,
essentially totally ionized. The H+ ions permeate the diaphragm and pass to the cathode cell under electrolysis.
2. At the cathode we have initially NaHSO4 ionizing mainly as Na+ & HSO4-. The electrolyte is acidic due to the further ionization HSO4- --> H+
+ SO4--, which has Ka2 ~ 1.2e-2 (compared with Ka1 ~10e3 for the first ionization H2SO4 --> H+ + HSO4-). The pH for a 1M solution will be around
2 due to this. The concentration [SO4--] is thus of order 10E-2 and the predominate negative ion is HSO4-. Both negative ions permeate the barrier
under electrolysis and increase the concentration of HSO4- in the anode cell.
The initial CE is thus about 97*3600/96500 = 3.66g/AH
3. As HSO4- ions are depleted from the cathode cell, more SO4-- ions are created there:
HSO4- --> SO4-- (to anode) + H+ (to cathode).
When the pH has reached ~ 7 (pure Na2SO4 is actually slightly alkaline due to the Ka2 ionization so pH of solution is then around 7.50 @ 1M ) all the
salt in the cathode cell can be considered as sulphate instead of bisulphate, [H+] being reduced to 1E-7. On continuation of electrolysis, [OH-]
increases to >1E-7 and competes for current with SO4-.
At this midpoint, 2F is needed to transfer one gram MW SO4-- ; twice as much charge as the initial rate, so the CE drops to 1.83 g H2SO4/AH.
4. Continuation of electrolysis increases the OH- cathode concentration until almost all SO4-- is transferred to anode cell; then obviously the CE
approaches zero and we are wasting current electrolyzing water to no useful purpose.
5. Hence if NaHSO4 is electrolyzed almost completely the overall CE will be <1.88g/AH. If Na2SO4 is the starting point, it will be <0.93g/AH; in
theory both are never complete, the end point being mainly water electrolysis due to OH- ion being greatly in excess of SO4-- ion.
Notice that nothing has been said about the anode cell concentration. If the above is correct, the only difference it would make is to the
overall cell conductivity. That means the anode acid concentration should not affect the CE. Thus it should be possible to concentrate to far
higher than the ~20% attained previously, provided adequate sulphate ions are present in the catholyte. Also, the only external manifestation
is that the cell is electrolyzing water to H2 and O2. Hence the amount of water lost should be estimable (by weight) as a guide to the total number of
Faradays passed through the cells
Finally, there are other options to increase CE, which I dismissed earlier on the grounds of ensuring there was very little sodium in
the acid produced, or constructional complexity. I may address these later.
Der Alte
|
|
|
Xenoid
National Hazard
Posts: 775
Registered: 14-6-2007
Location: Springs Junction, New Zealand
Member Is Offline
Mood: Comfortably Numb
|
|
Der Alte - greetings!
I came across the following electrolytic method for producing sulphuric acid. What is interesting is that it claims to produce concentrations as high
as 95%. The procedure is from "The Manufacture of Chemicals by Electrolysis" a treatise which appeared in 1919, and describes methods in use at the
turn of the century. The internet copy is confusing to read, has many mistakes and the diagrams are missing, but someone may be interested in trying
this out.
"ELECTROLYTIC SULPHURIC ACID is produced and a con-
centration as high as 95 per cent, obtained by oxidising
sulphurous acid in a diaphragm cell with a cylindrical
nickel cathode and an anode of platinum gauze. A porous
cup or cell which acts as cathode is filled with sulphuric
acid or sodium sulphite, and the outer anode compartment
contains a solution of sulphur dioxide which is kept
saturated during the process by passing in the gas
continuously. The current density used is about 1 amp. per
dm. 2"
I guess SO2 could be produced from a simple sulphur burner.
Use a platinum coated Ti mesh electrode.
May not be practical, but it seems to be an interesting amateur exercise! 
|
|
|
dann2
International Hazard
Posts: 1523
Registered: 31-1-2007
Member Is Offline
Mood: No Mood
|
|
Hello,
Just to add two cents worth for an alternative anode.
Would a Lead/Lead Dioxide plate from a car battery be better than a Lead only Anode.
Dann2
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
@ Dann2, greetings!
As far as the anodic oxidation of SO2 is concerned, yes, I have read that it has been done. I'm sure it could be done on a PbO2 anode. However, the
complexity of an efficient SO2 burner and gas transport problems make it a major task for easy amateur work. Yet adroit members here have made the
lead chamber process work, as you know.
I was looking to the day when the Nanny state legally forbids the sale of anything labelled acid! If you have H2SO4 then all others become fairly
easy to make, inorganic anyhow.
Would a Lead/Lead Dioxide plate from a car battery be better than a Lead only Anode? Probably - certainly a good PbO2 anode like those made by Swede
and yourself would! I just started from lead because even crudely oxidised the adherence is quite good, although thin, because the dioxide comes from
the lead itself (shades of Plante). You get quite a bit of flaking off. As a bonus, you get PbO2, no mean oxidant and interesting to play with.
I am considering making a deposited anode per your thread.
Also, currently, I am trying to come up with more efficient methods, current wise. I will report those soon.
Regards, Der Alte
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
I don't know how realistic this is, but could sulphur be oxidised directly by an oxidiser like ozone or H2O2.
A stirred suspension of sulphur in water should eventually form H2SO4 if ozonised air is bubbled through.
We know it works with HNO3!
|
|
|
Formatik
National Hazard
Posts: 927
Registered: 25-3-2008
Member Is Offline
Mood: equilibrium
|
|
| Quote: | Originally posted by hissingnoise
I don't know how realistic this is, but could sulphur be oxidised directly by an oxidiser like ozone or H2O2.
A stirred suspension of sulphur in water should eventually form H2SO4 if ozonised air is bubbled through.
We know it works with HNO3! |
Ozone appears to have no action on elemental sulfur (C.F. Schönbein, Lieb. Ann. 89 [1854] 257/300, 283), not even in the presence of H2O-steam (A.W.
Wright, Am. J. Sci. [3] 4 [1872] 29/31); but under certain conditions (these are not described in detail unfortunatley) there is oxidation to H2SO4
(C.F. Schönbein, Pogg. Ann. 67 [1846] 89/97, 92).
To add experiment, I've bubbled O3 in the form of ozonized air into aq. suspension of just gram amounts of sulfur flower. I noticed no visible
difference or significant action onto the sulfur bubbling ozonized air for IIRC over an hour. Were this to produce H2SO4, one would have to be
bubbling ozone in for hours and days. The results seem to reflect what is mentioned by the first reference above.
It would be better to burn to the sulfur and form SO2 and then oxidize or catalytically oxidize it. Oxidizing solutions is also in order, it should be
simple enough to use ozone to oxidize aq. H2SO3 solutions to H2SO4. But this all has nothing to do with electrolytic methods.
[Edited on 4-2-2009 by Formatik]
|
|
|
chief
National Hazard
Posts: 630
Registered: 19-7-2007
Member Is Offline
Mood: No Mood
|
|
SO2-oxidation on the anode: Why not try it using Na2S2O5, which is readily available for food-chemical uses ? 1 kg == 1 $, at 25 kg=bags ... ; also in
smaller amounts available at the hardware-store, for disinfection of home-made wine etc. .
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Further ideas to increase current efficiency (CE)
If we waive the condition of very little sodium in the acid produced, we can start with NaHSO4 in both anode and cathode cells. Then
initially, on electrolysis, the ions passing through the diaphragm will be chiefly Na+ from anode side to cathode, and HSO4- in the reverse direction
(ignoring the much smaller SO4-- ion concentration). Simultaneous enrichment of HSO4- and depletion of Na+ will occur at the anode cell. 1 Faraday
will move ~1 mol Na+ (leaving behind 1 mol HSO4-, and will also move ~1 mol HSO4- into the anode cell, for a total of 2 mol HSO4- per Faraday not
neutralized by Na+. Initial CE, g/AH of increase in unneutralized SO4 concentration, twice the previous amount or about 7.3g/Ah.
The general condition while both cells are acidic can be shown as follows, (brackets = relative amount for pH~1 in both cells):
Anode cell to cathode cell: <-- Na+ (major); <-- H+ (minor) …. (1).
Cathode cell to anode cell: --> OH-(very small); -->SO4-(minor); -->HSO4- (major) ..(2)
At cathode: 2H+ + 2e- --> H2(g)
At anode: 2HSO4- --> 2HSO4* + 2e-; HSO4* + H2O --> H+ + SO4- + ½ O2(g)
* = transient radical.
As the anode cell increases its acid content its pH falls; further, as Na+ ions are abstracted [Na+] decreases. Hence the efficiency of the Na+
transfer decreases as [H+] increases. Simultaneously, the [HSO4-] drops in the cathode cell and [SO4--] increases, and the transfer of SO4 moiety
decreases for a given current.
Once the cathode cell approaches pH 7, the efficiency of SO4 transfer has dropped to 50% and further increases in pH cause it to drop much lower, due
to increasing [OH-]. At the anode pH decreases further, [H+] increases and the transfer rate of Na+ to the cathode cell diminishes. It is thus
theoretically impossible to deplete the anode cell entirely of Na+ ions.
I have attempted some rough calculations to see what this variation of efficiency might be theoreticlly. We are using very concentrated solutions so
the accuracy is likely to be very approximate, due to variations in ionic activity, but such calculations show a trend. I used a pH calculator to
estimate the ionic activities, but although there is some correction, the concentrations are way outside the range of accurate results. I will not go
into detail because the calculation proved long and tedious. It does not help at all that sulphuric acid is diprotic!
The results can be applied to the original case discussed previously, where there are no Na+ ions in the anode cell (well, very few.)
The best presentation seems to be graphical. The key parameter is the ratio of [Na+] to [total SO4], i.e. or alternatively [H+] or pH. The dependent
quantity is the efficiency as compared to one mol HSO- per Faraday = 3.66g H2SO4/Ah. A separate graph of pH vs. [SO4--] +[HSO4-] is also shown.
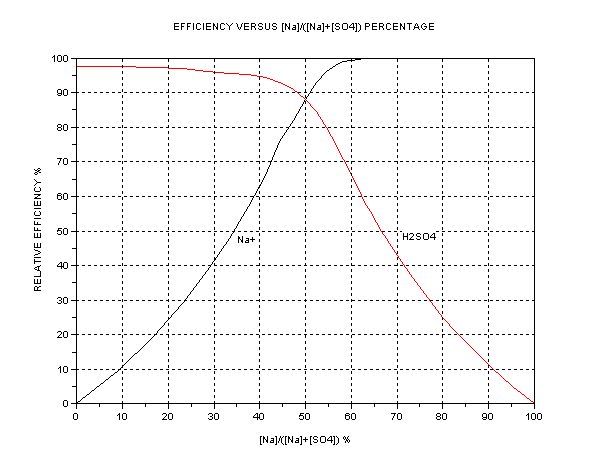
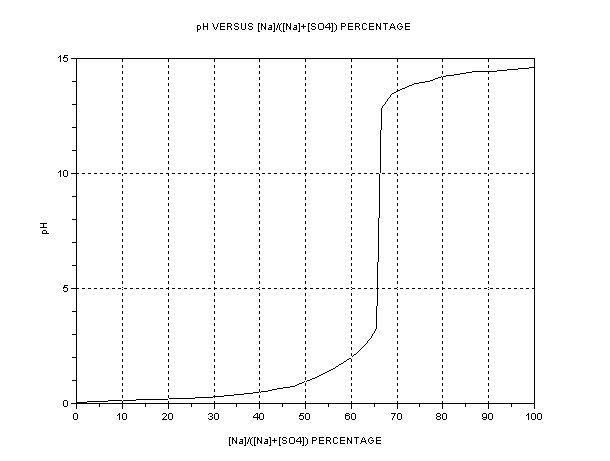
The efficiency of Na+ transfer from anode cell (A) to cathode cell (C) is
100*[Na+]/{[Na+]+[H+]}
and of SO4 ions C --> A is
100*{[HSO4-] + [SO4--]}/{[HSO4-] + 2[SO4--] + [OH-]}
The pH refers to solutions in the region of 2-3 M where ion activity tends to be variable, typically. Accuracy not to be expected except near the
critical neutrality point. If it looks like a traditional titration curve, it is – the only difference is that only concentrations vary instead of
concentration and volume.
This graphs indicate that in the case where cathode cell C contains NaHSO4 initially (50% Na to SO4 ion ratio) the expected efficiency is about 88% of
3.66 = 3.22 g/Ah. (The commercial NaHSO4 usually contain 8% Na2SO4 and this drops the efficiency to about 3.1g/Ah). This accords closely with what I
measured initially earlier. When the C contents become Na2SO4 (neutral, at 66.7% Na) this efficiency drops to 50% exactly = 1.83gAh. Further
electrolysis gives diminishing returns; at 90% Na it is only 0.4g/Ah. If the aim were to produce pure NaOH in C, it would take forever. Hence an
indicator or pH meter is a help if you want to keep efficiency high. The ‘efficient’ region is narrow, between 50 to 66.7% Na/SO4 ratio. This
means only ½ of the sulphate in NaHSO4 can be transferred efficiently from C to A.
If both A and C are started with NaHSO4, as much Na is transferred to C as SO4 to A initially. This results in about twice the above rate of
enrichment of H2SO4 in A, i.e. about 6.4g/Ah. As C becomes less acidic, the rate drops as before. When C becomes neutral, this rate is 50% of initial
Na to C from A, 1.83g/Ah. The C to A transfer rate of SO4 moiety depends on the relative C concentrations, and the relative size of A and C. It helps
to have a large cathode cell C to avoid this dropping rapidly – a coaxial cell usually guarantees the volume of C > volume of A.
I have tried this method. The expected efficiencies are indeed much higher, but of course you still have some sodium in the acid produced.
In the case of only acid in both cells (concentration mode, as suggested by chloric above), relative efficiencies of about 98% ( ~3.5
g/Ah) would be expected (no Na+). This method can also be used to get rid of residual sodium efficiently. However, the overall process efficiency
drops to about the 1.8 g/Ah for both processes in sequence because the electrolytic process is used twice.
There is no bar to producing concentrated acid, at least up to 45%, I have found in my latest experiments.
One other experiment is worth mentioning, even though it proved unproductive.
Consider a 3 part coaxial cell. The anode cell contains the PbO2 anode; the center cell saturated NaHSO4; cathode could be Fe or carbon, Ni, etc.
Anode cell would be primed with H2SO4, very dilute; cathode with NaOH, very dilute, the purpose being to increase initial conductivity. The starting
NaHSO4 concentration is about 4.8+ M in the center cell. On electrolysis the following should occur (arrows show flow direction.:
Cathode cell from center cell: <--Na+ (mainly); <-- H+ (minor)
Cathode cell to center cell: --> OH-
Anode cell from center cell: --> HSO4- (mainly); --> SO4-- (minor)
Anode cell to center cell: <-- H+
In center cell, H+ + OH- --> H2O (controlled by pH level)
At cathode: 2H+ + 2e- <-- H2(g)
At anode: 2HSO4- --> 2HSO4* + 2e-; HSO4* + H2O --> H+ + SO4- + ½ O2(g)
* = transient radical.
At the center to cathode barrier: OH- + H+ --> H2O
In words, the NaHSO4 in the center cell is depleted equally of Na+ and HSO4- ions while at the same time water is generated in the center. This
produces, ideally, a solution of H2SO4 at the anode and one of NaOH at the cathode, pure, simultaneously.
The expected maximum CE for this process is ~1 mol/F of HSO4- or Na+ transferred, i.e. the ~3.2 g/Ah level of H2SO4. This rate will not decrease much
until the pH goes above about 2, when SO4- - and H+ ions begin to predominate in the center cell. This can be postponed by adding crystalline NaHSO4
to the center cell, which is slowly diluted by water @ ~1g/Ah. This CE would be 7 times greater than the average 0.4g/Ah got in the previous post.
I tried this. It works but the overall resistance of the cell was surprisingly high, 0.5 amp for 16V impressed, and that for over 400 cm^2 of center
pot surface. This was traced to a large drop across the center/cathode barrier. My guess is that the water produced by the last reaction above is
creating a layer in the 5mm thick terracotta pot that is essentially pure and non conductive. The current efficiency was about as expected but the
power efficiency abysmal. At the current obtainable it takes forever to get any acid and alkali. So another good idea bit the dust! If my theory of
water production is correct, a much thinner diaphragm should help to reduce this dreadful power inefficiency.
A Few Experimental Observations
The NaHSO4/H2SO4 method, outlined previously, was repeated. Essentially the same results were obtained as above, except the electrolysis was stopped
once the cathode cell became alkaline. The anode contents were then used to start off a new electrolysis, the NaHSO4 cathode level being renewed.
43.5% acid was obtained in this way at fair efficiency, around 2g/Ah.
The two cell NaOH/NaHSO4/H2SO4 version was run for some time at the rather low current level of ~0.5 amp. Titration of the NaOH against roughly
standardized HCl showed that the resultant cathode solution was about 0.8N, the anode content being about 2M H2SO4. The efficiency was > 1.5g/Ah,
but difficult to estimate due to the changing nature of the current under severe temperature fluctuations. (Run outside on one of our rare cold
nights.)
The NaHSO4/NaHSO4 2-cell method produced acid at relatively high efficiency, difficult to estimate because of remaining Na+ content. One interesting
point was that persulphate and/or H2O2 was also produced. The acid bleached the indicator I used (phenol red). The resultant anode liquor produced
bubbles on standing, suspected of being oxygen but not proven. This acid could, but has not yet been, purified of sodium and further concentrated by
electrolysis in a H2SO4/H2SO4 cell.
I now have over a kg of Na2SO4. It crystallizes in huge crystals as Na2SO4.10H2O if done slowly – some I got were about 10 cm by 1.5cm, prismatic.
The solution supersaturates with great ease. If rapidly cooled, a single crystal added will cause the entire precipitation in a few seconds. You can
see the crystals growing as long needles. It is not possible to keep these crystals unless the relative humidity is > ~ 50% - they effloresce. And
at 32C they melt, or dissolve in their water of crystallization. Since NaHSO4 is far more soluble, the two salts can be separated this way and the
bisulphate returned for electrolysis.
NaHSO4 also forms large crystals as monohydrate, but these are quite different in shape, being compact and not needles. I was not able to get a
perfect crystal, but they are monoclinic or rhomboidal, not far from cubic, as far as I could judge.
@chief
WRT Sulphites, etc. :- I think sulphites or metabisulpites (like Na2S2O5) would work in a divided cell, but poorly. H2SO3 is a weak acid (Ka1 = 1.89
, Ka2 = 7.21). In the strong acid environment of the anode cell, the SO3- - ions entering would tend to become un-ionized H2SO3 and their
concentration would be low compared with the HSO4-. SO2 might be evolved. Anodic oxidation only takes place on the ions, so efficiency would be poor.
In an undivided cell sulphate would result. However, SO2 dissolved in an undivided cell, especially if bubbled over the anode, should work.
Thiosulphates, and the various thionates and thionites also would do something, but possibly also produce elemental sulphur. NaHSO4, being an
industrial byproduct, is very cheap; I’m sure it must be cheaper than Na2S2O5.
Regards, Der Alte
|
|
|
| Pages:
1
2 |
|











