| Pages:
1
2
3 |
stoichiometric_steve
National Hazard

Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | | Originally posted by KluteHave you considered the Ti isoproxide route? The "regular" grade isn't that expensive, and the reaction is said to
work well with delicate substrates... |
if that route is attractive as the papers state it, throw away the STAB, mol sieves and so on.
i've even seen the use of regular TiO2 in pressurized catalytic hydrogenation of amines with carbonyl compounds (some german patent). i think that the
Ti(IV) has a mediating effect on the imine formation.
check this btw.: Reductive Amination with Ti(OIPr4) and Pd/C without solvent
[Edited on 29-3-2008 by stoichiometric_steve]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Nice paper! I had only stumbled on the ones where NaBH4 is used as reducer. Obviously, once the titanium-imine complex is formed, it could be reduced
in several ways. This leads me to ask myself if CTH couldn't be used here? This would greatly simplify the procedure, and diminish the cost
(recycleable catalyst).
The way i see it, the titanium complexes the imine, so it isn't an equilibrium anymore and there's noketone to be reduced.. Even less than equimolar
are said to be usefull when the reducing agent is gradually added, as the Ti complex get's reduced very quickly, and the liberated Ti IV reforms a
complex with the remaining ketone and amine, etc etc.
If you can't get the Ti(OiPr)4 for a reasonable price, go for it. When ever I'll think of doing a reductive amination, i'll use it for sure! I can
have it for 17.43E 100mL, which is much cheaper than pur STAB or 4x NaBH4 and certainly less that NaBH3CN!
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
Very nice paper indeed......I wonder then if the amine can be the salt hence solving a headache in the reductive amination of L-PAC.......solo
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by solo
Very nice paper indeed......I wonder then if the amine can be the salt hence solving a headache in the reductive amination of L-PAC.......solo
|
solo, as stated in the other papers (i mean this one), the authors use Ammonium Chloride and Methylammonium Chloride along with Triethylamine (ratios: Substrate:Amine
Salt:Triethylamine:Ti(OIPr)4:NaBH4 1:2:2:2:1.5) stirred for ~10hrs, then adding NaBH4 and stirring for 8hrs.
so, here's the solution to your problems 
Klute, where did you read about the subequimolar amounts of Ti(OIPr)4 being used? i'd love to see that, since the Ti(OIPr)4 isnt really that cheap
(2kg=7.5mol=150EUR)
[Edited on 29-3-2008 by stoichiometric_steve]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
In the paper you provided, table 1 last entry, and i could have sweared i saw it in one of pdf already mentionned, but it doesn't seem to be the
case.. I have a look elsewhere.
|
|
|
Methyl.Magic
Hazard to Others
Posts: 139
Registered: 14-5-2007
Member Is Offline
Mood: No Mood
|
|
| Quote: | Message original : grind
You can add 4 or more moles of AcOH to 1 mol of NaBH4, in the cold only triacetoxyborohydride is formed. The last H is replaced at elevated
temperatures with an excess of acid.
You need very finely powdered NaBH4 and the complete conversion to triacetoxyborohydride takes a lot of time (min. 12 h) in the cold, even with an
excess of AcOH. The suspension becomes gel-like during the process.
[Edited on 27-3-2008 by grind] |
Thank you !
This is important to know ! I was wondering if it was like the preparation of LiAlH(tBuO3) (dropping LiAlH4 in t-butanol).
|
|
|
grind
Hazard to Others
Posts: 120
Registered: 13-1-2007
Member Is Offline
Mood: No Mood
|
|
4,29 g of finely powdered NaBH4 (assay 96%, 0,11 moles) is dried at 140°C in vacuum for 1 hour. It is then suspended in 68 g of dry toluene and the
lumps are broken again if necessary. The suspension is cooled with an ice/NaCl bath and a mixture of 27,55 g glacial AcOH (dried with Ac2O, 0,46
moles) and 70 g of dry toluene is added within 70 minutes (exclusion of moisture and stirring with a very large magnetic stirring bar at approx. 400
rpm during the whole process). The cooling bath is removed 1 hour after the addition of the AcOH/toluene mixture was finished. Now the suspension is
stirred for approx. 12 hours at room temperature or until the evolution of H2 virtually ceased. The thick, gel-like white suspension (which is now
more difficult to stir) is now ready for use. You can add a ketone/amine mixture or whatever you want. The progress of the reaction seems to help to
destroy the gel-like state of the mixture, and stirring becomes easy again.
This is my personal method and experience to make STAB. The amounts of toluene can be varied widely.
Sorry for my bad english.....
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
In addition to Grind's excellent preparation, I would like to add this preparation of STAB I found randomly. The procedure is pretty similar, except
that they isolate the STAB as a powder to be stored before use, and use benzene instead of toluene...
| Quote: |
Sodium Triacetoxyborohydride. A 100” Schlenk flask equipped
with a Schlenk filter was charged with 186 mg (4.93 mmol) of sodium
borohydride and 50 mL of anhydrous benzene. The slurry was cooled
to 10 OC and 860 pL (15.0 mmol, 3.04 equiv) of anhydrous acetic acid
was added dropwise so as to maintain an internal temperature no higher
than 20 OC. Hydrogen evolution was measured with a gas buret. After
addition of acetic acid was complete the mixture was allowed to warm
to ambient temperature and stirred at that temperature for 8 h. Hydrogen
evolution had ceased at 330 mL (theoretical for 3.00 equiv is 331
mL) after 5 h. The colorless slurry was filtered and the resultant white
powder washed with three 20-mL portions of freshly distilled, anhydrous
ether. The combined ether filtrates did not liberate hydrogen when
treated with 1 N aqueous hydrochloric acid. The powder was held under
vacuum over night to afford 961 mg (92%) of analytically pure sodium
triacetoxyborohydride as a white, hygroscopic solid:
|
Article
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by stoichiometric_steve
if that route (Ti(OiPr)4) is attractive as the papers state it |
gotta correct myself here. i tried the method, and it is awful. lots of salts at the end of the workup, product sticking to the salt, unfilterable,
messy. unfeasible unless steam distillation is suitable for product separation.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Here is a pretty inspiring procedure for the reduction of an enamine with preformed acetoxyborohydride species in excess GAA (required to form the
iminium salt that can then be reduced).
In this example, they use an amine-borane complex, but borohydrides (NaBH4, NaBH3CN, etc) are equally used (results in the tables) with often
excellent yields.
This in-situ preparation of STAB in excess GAA simplifies the procedure, and apparently avoids the thick gel-like suspension. They used GAA straight
from the bottle.
| Quote: |
Reduction of 4-tert -Butyl-1-( 1-pyrro1idinyl)cyclohexene with (CH3)3CNH2BH3 (Reverse).
The amine-borane (0.87 g, 10 mmol) was stirred with 30 mL of glacial acetic acid under Ar for 12 h, the enamine (1.04 g, 5 mmol) was then added, and
the solution was stirred at room temperature for 16 h. Concentrated HCl (4 mL,) was added, and the solution was stirred for 1 h, poured onto ice, and
made basic with 50% aqueous NaOH. The mixture was thoroughly extracted with ether, and the organic phase was dried (K2CO3), concentrated after GC
analysis (Table VII), and flash distilled on a Kugelrohr apparatus to afford 0.93g (89%) of the cis- and
trans-N-(4-tert-butylcyclohexyl)-pyrrolidines.
GC analysis indicated an 87/13 cis/trans ratio (Table
11). For examples not conducted under reverse conditions (Tables I and III), the above procedure was modified only in that the enamine was dissolved
in acetic acid followed by the amine-borane.
|
From: Stereoselective reductions of substituted cyclohexyl and cyclopentyl carbon-nitrogen .pi. systems with hydride reagents
J. Org. Chem.; 48(20); 3412-3422 (1983)
(availble in the ref's forum thanks to Vovan78)
In the reversed conditions, acetoxyborohydrides are not the actual reducting agents, the borane complex is (or resp. the borohydrides). This kind of
addition is used in:
Reduction of Steroidal Enamines
James A. Marshall and William S. Johnson
J. Org. Chem.; 28(2); 421-423 (1963)
(avaible in the ref forum too)
| Quote: |
3p-N-Pyrrolidylcholestene-5 (IIb). (a) Sodium Borohydride
and Acetic Acid Reduction.-A mixture of 50 mg. of 3-
N-pyrrolidylcholestadiene-3,5,m~ .p. 136-141", and 25 mg. of
sodium borohydride in 1 .5 ml. of anhydrous tetrahydrofuran
was stirred under nitrogen, and 0.5 ml. of glacial acetic acid was added dropwise. A 0.5-ml. aliquot was removed, made basic with 10% sodium
hydroxide, and extracted with ether-ethyl acetate.
The extracts were washed with water, saturated brine,
and dried over anhydrous granular sodium sulfate. Removal
of solvent left 14 mg. of colorless [amine-borane complex]
The remainder of the reaction mixture was refluxed for 1 hr.,
and the product was isolated as above to give 36 mg. of crude solid.
Recrystallization from ethanol afforded 16 nig. (44% yield allowing for removal of the aliquot) of lustrous white plates, m.p.165-175O.
The infrared spectrum of this material was identical
with that of material prepared as described below (part b).
Admixture of the two substances gave no depression in melting
point.
The utilization of diglyme as the solvent led to a substantial
improvement in yield. Thus, a solution of 50 mg. of 3-Npyrrolidyl~holestadiene-3,5,m~ .p. 136-141 ", and 20 mg. of
sodium borohydride in 2.0 ml. of anhydrous diglyme was stirred
under nitrogen during the dropwise addition of 2.0 ml. of glacial
acetic acid. The mixture was allowed to stand for 10 min. and
heated on a steam bath for 1 hr. The crude product was isolated as described above and crystallized from ethanol to give 30 mg. (60% yield) of
3@-N-pyrrolidylcholestene-5, m.p, 160-172'. The mother liquors afforded 17 mg. (34% yield) of an
oil, the infrared spectrum of which was nearly identical with
that of crystalline amine IIb.
|
Hope it can be usefull to some (Chemrox?).
I plan on trying a few similar reductions, and will report my results when done.
EDIT: I have one question for anyone with experience with formation of STAB, does the sue of THF as cosolvent in the preparation (Grind's procedure)
give a gel-like sludge too? Or is it possible that a solution is formed?
there is a mention of the preparation of the STAB by addition of GAA to a susepsnion of NaBH4 and subsequent transfert of the "solution" to the
imine/enamine, but it's on a pretty large scale and laking of details, so it could just aswell be a suspension or a slurry.....
[Edited on 22-8-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by stoichiometric_steve
gotta correct myself here. i tried the method, and it is awful. |
gotta correct myself once more! the method is beautiful! sticking to the instructions is the way. the workup needs HUGE amounts of water/dilute acid
or dilute base to generate a filterable solid. approximately 1 - 1.5L final volume for a 300mmol run.
|
|
|
akdovnoff
Harmless
Posts: 1
Registered: 26-6-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Klute
EDIT: I have one question for anyone with experience with formation of STAB, does the sue of THF as cosolvent in the preparation (Grind's procedure)
give a gel-like sludge too? Or is it possible that a solution is formed? |
Although I don't have experience with the actual preparation of STAB, I've used it quite a lot and it's pretty much insoluble in THF. Infact, it's
solubility in pretty much anything is distinctly limited from what I've seem.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thank you very much for that information. I guess i will go with in-situ preparation in GAA... Could you comment on the stability of this reagent
towards atmiospheric humidity? Is it as sensitive as NaBH4, or does it really require handling in dry box?
I'm considering preparing 100-200mmol and isolate it as a solid, and was wondering if regular glass frit would be ok for filtration, or if schlenk
filtration was required....
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I have run a STAB reduction, preforming it in situ by adding NaBH4 to 10x molar GAA, follwing a procedure for enamine reduction (J. Org. Chem., 48,
3412-3422 (1983)). I was hoping to obtain a fluid solution of STAB in GAA.
So 3eq of NaBH4 were added to 10x molar amount of GAA over 30min, at ice bath temperature (5% anhydrous toluene was added to avoid cristn of GAA at
those temps), in small portions, keeping the temp at 0-5°C.
The reaction is pretty exothermic, so small portions must be added, when the vigorous H2 evolution calms down. Near the end of the addition, the
bubbling mixture suddenly turned to a thick, white paste, nearly impossible to stir. Anhydrous toluene was added to thin it up, obtaining a fluid, gel
like suspension, as per Grind's description. The suspension was left to stir for another 30min, at which point the H2 evolution had virtually stopped.
The enamine was added, followed by some toluene to thin up the slurry formed, and the mixture stirred for 24H.
After hydrolysis of the boron complex by stirring with HCl for 30min, which formed a thick emulsion of fine white solids, basic workup worked like a
charm, dissolving the fine emulsion-creating white boron solids. After a complete A/B, the amine was obtained in ~70% yield.
So, basicly, there is no point in pre-forming STAB this way: addition of NaBH4 portion-wise causes unneccesary exposition to atmospheric moisture, and
is much less practical than adding GAA dropwise to NaBH4 suspended in toluene, or other solvent. A co-solvent being needed during the formation of the
hydride and the reduction to obtain a stirreable mixture, it is much more practical to add the substrate dissolved in an anhydrous co-solvent.
The yield was pretty satisfying, but having discovered that the use of Zn dust in GAA is as effective with aromatic enamines, there isn't much point
is "wasting" NaBH4 (3 hydrides eq. going off as H2).
I have a few pictures if people are interested. If I try Grind's procedure I will report back.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
I found the reading of this to be most interesting and rewarding.......solo
One-pot reductive amination of aldehydes and ketones with α-picoline-borane in methanol, in water, and in neat conditions
Shinya Sato, Takeshi Sakamoto, Etsuko Miyazawa, Yasuo Kikugawa
TetrahedronVolume 60, Issue 36, 30 August 2004, Pages 7899-7906
Abstract
A one-pot reductive amination of aldehydes and ketones with amines using a-picoline-borane as a reducing agent is described.
The reaction has been carried out in MeOH, in H2O, and in neat conditions in the presence of small amounts of AcOH. This is a highly
efficient and mild procedure that is applicable for a wide variety of substrates. In particular, this is the first successful
demonstration that this
type of reaction can be carried out in water and in neat conditions.
---------------------------------------------------------------------------
Attachment: One-pot reductive amination of aldehydes and ketones with α-picoline-borane in methanol, in water, and in neat cond (219kB)
This file has been downloaded 1516 times
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
That's a pretty impressive and promising paper!
Considering the aq. solution, it would seem that amine-borane complexes are selective enough to reduce even traces of imines/enamines without touching
carbonyls... Also, the su eof only 1eq of amine si pretty interesting.
I suppose pic-BH3 might not be too difficult to produce from NaBH4 and a lewis acid BF3.Et2O for example, generating dibornae and complexing it with
the amine.
On the other ahnd, picolines aren't such common reagents, so maybe BH3.trimethylamine or BH3-pyridine can substitute well enough with simialr results.
The drawbacks from using pyr-BH3 mainly concern industrial scale...
The procedure I used from the above enamine reduction actually uses t-BuNH2-BH3 in the experimental, so it seems pretty much similar. The authors
claim that the borane forms an acetoxyborane species in-situ before adding the substrate, when adding the hydride before the substrate in GAA.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
I think that if the catalyst is made easily this will replace the very toxic HgCl and aluminum type reductions and also the sodium borohydride and
catalytic hydrogenations in the near future.....will run a reaction to see how it works and it's yield and will report........solo
Here is some other stuff on boranes,
http://www.organic-chemistry.org/chemicals/reductions/borane...
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
On a side note, production of borane from various Lewis acids, for amide reductions (Kindly found by Nicodem):
Enhancing Borohydride's Reductive Selectivity
Apparently, borane-amine complex are generally prepared by passing a stream of anhydrous CO2 in a mixture of NaBH4, amine, and solvent, in dry
conditions (US 5,144,032).
But there is also a very promising method: reacting the amine hydrochloride with NaBH4 in ethers: (Coll. Czech. Chem. Commun. 34, 3009 (1969) )! The
inorganic salts are filtered off, and the solvent evaporated to yield the amine-borane complex!
US H000919 present the preparation of tertiary diamines-borane complexes with CO2 or GAA, and reviews several other patents. Several methods
really seem interesting. The triethylamine-borane complex seems like a good candidiate too.
I think we have something here! very nice find Solo!
edit: This article seems pretty interesting, it suggests that the formation of ammonium borohydrides by adding amine salts to NaBH4 maybe a first step
in reductive aminations...
This kind of gives me a idea for really easy redutive amination: to use the hydrochloride of the primary amine, add a more basic tertiary amine
(Et3N), forming the hydrochloride of the tertiary and the freebase of the priamry, adding 1eq of NaBH4, forming the triethylamine-borane complex in
presence of the primary freebase, and adding the ketone/aldehdye, forming the imine with the primary, which is reduced by the amine-borane complex....
I don't think this can work with pyridine, might no be basic enough to liberate the primary amine.
I suppose the NaBH4 will react with the acidic ammonium salt, forming ammonium borohydride, which decomposes to the amine-borane complex. But we need
to be sure the tertitary amine complex is more stable than the primary-amine one, to avoid some kind of borane transfert, consuming the primary amine.
But I suppose the possible aq. conditions could allwo us to simply avoid triehtylamine, use commercial aq. solutions of the primary amines, or in-situ
from the ammonium salts and hydroxides: generated water doesn't seem to be a problem.
The questions would then be how availble the picolines are...
[Edited on 4-9-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
grind
Hazard to Others
Posts: 120
Registered: 13-1-2007
Member Is Offline
Mood: No Mood
|
|
| Quote: | | Originally posted by KluteI have a few pictures if people are interested. |
I´m interested in your pictures .
If you try "my" procedure, you can use much more toluene of course to make the mixture easier to stir.
Also you can use THF or 1,2-dichloroethane as solvent.
There is an article where they use propionic acid and THF and when I remember correctly, they get a solution.
Acids with higher molecular weight than acetic acid give a higher stereoselectivity in the reductions.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Here goes:
- The setup (oven dried, cooled under Ar)

-The enamine (aniline and N,N-diethylbenzoylacetamide in toluene, cat. TsOH and Dean Stark, removal of most of the toluene):

-NaBH4 (dried over KOH overnight):

-GAA+ anhydrous toluene (3A MS), added via syringe:

-Paste obtained near the end of the addition:

-After adding anhydrous toluene:

-Addition of the enamine:
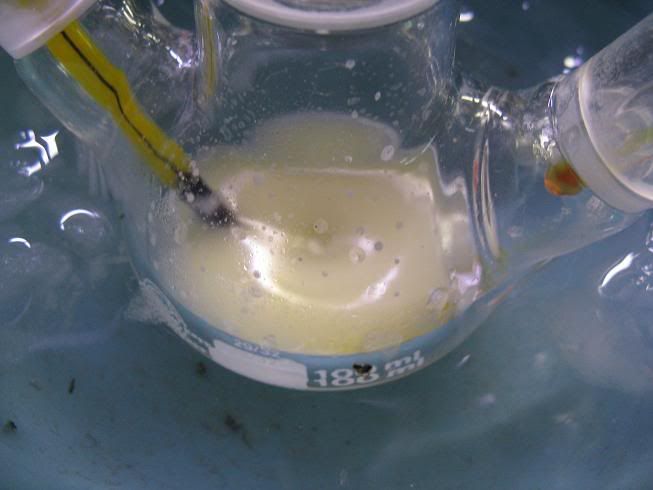
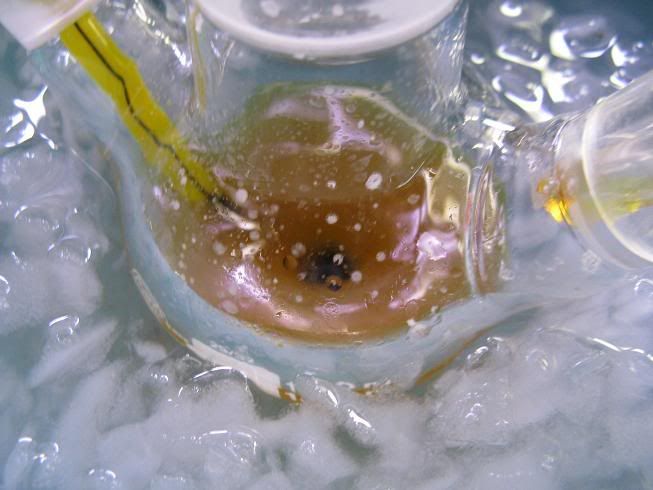

-After 24H stirring at RT (notice fine cristalline solid):

-After addition of conc HCl dropwise (quench of the exces hydride, and decomposiiton of the amine-borane complex):

Didn't take any more pictures after.... After stirring at RT for 30min, adding conc. NaOH until most of the solids/emulsion had dissolved gave a
clear aq. and dark orange/brown organic layer, which was washed with water and extracted with dilute HCl, aq. washed with toluene, basified and amine
extracted, washed, dried, and cristillized from HCl/IPA in ~70% yield.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
grind
Hazard to Others
Posts: 120
Registered: 13-1-2007
Member Is Offline
Mood: No Mood
|
|
Thanks for the pics, Klute. Good work and very interesting!
Did you make an enamine from an amide (N,N-diethylbenzoylacetamide) and aniline? What´s the structure of your carbonyl compound?
Was the suspension easier to stir after the addition of the enamine solution?
Quenching directly with conc. HCl is dangerous. Borane can be formed and cause fire/explosions. A safe way is quenching with water or NaOH-solution.
I read in an article about NaBH4 that it loses its water above 35°C. So I guess drying with KOH is not as efficient as drying at elevated
temperatures. I dried it at 150°C in vacuum for 1 h and there was a little loss in weight (12,04 g ---> 11,91 g).
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by grind
Quenching directly with conc. HCl is dangerous. Borane can be formed and cause fire/explosions. |
Where did you read/hear about that? i usually use 20% aq. H2SO4 or straight 99% GAA to quench even NaBH4 powder, i never ever had a fire. In aqueous
solution, Borane is never going to be around in gaseous form to cause trouble.
|
|
|
grind
Hazard to Others
Posts: 120
Registered: 13-1-2007
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by stoichiometric_steve
| Quote: | Originally posted by grind
Quenching directly with conc. HCl is dangerous. Borane can be formed and cause fire/explosions. |
Where did you read/hear about that? i usually use 20% aq. H2SO4 or straight 99% GAA to quench even NaBH4 powder, i never ever had a fire. In aqueous
solution, Borane is never going to be around in gaseous form to cause trouble. |
Yes, in aqueous solution it´s no problem. So it´s better to first add water and then acid, even when you work with diluted acid. I´ve often read
this in the literature and there were such accidents reported.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Here is the original procedure, where they use a borane -t-BuNH2 complex as hydride:
| Quote: |
Reduction of 4-tert -Butyl-1-( 1-pyrro1idinyl)cyclohexene
The amine-borane (0.87 g,
10 mmol) was stirred with 30 mL of glacial acetic acid under Ar
for 12 h, the enamine (1.04 g, 5 mmol) was then added, and the
solution was stirred at room temperature for 16 h. Concentrated
HCl (4 "1,) was added, and the solution was stirred for 1 h,
poured onto ice, and made basic with 50% aqueous NaOH. The
mixture was thoroughly extracted with ether, and the organic
phase was dried (KzCO3), concentrated after GC analysis (Table
VIT), and flash distilled on a Kugelrohr apparatus to afford 0.93
g (89%) of the cis- and trans-N-(4-tert-butylcyclohexyl)-
pyrrolidines. GC analysis indicated an 87/13 cis/trans ratio (Table
11) |
Indeed, I think adding a little water first can't do any harm. The reaction with STAB will be a little less vigorous I suppose.
The carbonyl compound I used is the N,Ndiethylamide of benzoylacetic acid, made by reacting excess ethyl benzoylacetate and diethylamine in presence of cat. NaH at high temps and subsequent removal of ethanol formed.
One could think that with a primary amine and a ketone an imine would form, but b-ketoester and b-ketoamides always form stable enamines, the double
bond formed being conjugated with the carboxyl. Apparently, it isn't just a mixture of enamine and imine tautomer, but only the enamine at all times.
Hence, the reduction must be run with 1 eq. of GAA or simialr to form the iminium salt...
Yes, the slurry was definitively easier to stir after adding the enamine, but I did add at least an equal volume of toluene (the imine took a few
seconds to correctly dilute itself in the mixture, while in solution it was instantaneous). There was a little H2 evolution at first, but it quickly
came to a stop.
I only hav acces to a kitchen oven at home, and am very reluctant at putting chemicals in it  The KOH seems good for my purposes, I was thinking of using P2O5, but I prefer keeping non-acidic conditions to avoid any H2 evolution if
a grain of NabH4 falls on the P2O5 for some reason... The KOH seems good for my purposes, I was thinking of using P2O5, but I prefer keeping non-acidic conditions to avoid any H2 evolution if
a grain of NabH4 falls on the P2O5 for some reason...
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
grind
Hazard to Others
Posts: 120
Registered: 13-1-2007
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Klute
I only hav acces to a kitchen oven at home, and am very reluctant at putting chemicals in it The KOH seems good for my purposes, I was thinking of using P2O5, but I prefer keeping non-acidic conditions to avoid any H2 evolution if
a grain of NabH4 falls on the P2O5 for some reason... |
You don´t need an oven, drying directly in the reaction flask not only removes water traces in the NaBH4, but also makes your glassware perfectly dry
for the reaction. Especially if you powder the NaBH4 in the air, it becomes wet due to its hygroscopic nature. Heating it after the powdering
procedure removes water traces reliably.
The use of argon is not necessary in the most cases, except your imine/enamine is very air sensitive. The exclusion of moisture is much more
important, otherwise, even reductions with aqueous formaldehyde solution were performed with a large excess of STAB.
|
|
|
| Pages:
1
2
3 |









