| Pages:
1
2
3
4
5 |
Klute
International Hazard

Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Well, the TAA.HSO4 isolated from the methanol seems definatively free of ammonium salts, as when basified no ammonia smell was detectable (there could
haev been minutes amounts covered by the smellof TAA).
It was just too tedious as the fine solid remaining was very fine and was long to decant, so I just did one extraction with ~150mL MeOH (recovered
less than 100mL), which furnished the 6.9g I used for the oxidations.
I'm sure if I had continued these extractions I would have recovered more TAA. When I dissolved the solid, i used too much water: the formed TAA
hydrate must have stayed solubilized in the water, or partially precipitated with the massive amount of Na2SO4.10H2O and other salts, hence the smell
and the colour change with the marquis.
I'm going to try more basic methods like refluxing concentrated ammonia with acetone and ammonium salts and stick to that, satrting to get bored of
using ammonia gas, I would be happier if the recation could take care of itself while I do other things.. I have still the MgO/CaO run to work up,
but that was one a rather small scale any way.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
I'll grab some benzoate salts and try the ammonium benzoate method, at least I should get something
Here is the other ammonium salt article: http://www.biochemj.org/bj/027/0157/0270157.pdf
Apparently ammonium benzoate gives predominantly the TAA-benzoate when refluxed with acetone IIRC, I have to look the article up again
BTW Here is something of interest: http://www.rsc.org/delivery/_ArticleLinking/DisplayArticleFo...
See page 18/60 where it describes the effect of adding alkali metal alcoholates to triacetoneamine, ie. the formation of the alkali metal salt (by one
Emanuel Merck no less). Now I seem to remember reading somewhere that dry alcohols mixed with alkali metal hydroxides exist predominantly as alcoholic
solutions of the alkali metal alcoholate? Would this explain why this is acting weird?
Whhhoooppps, that sure didn't work
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Was the ammonium benzoate used stoechiometrically or catalytically? If used insteochiometric amounts, hopefully a less expensiveve salt can be used..
I have a little by-product benzoic acid at hand, i could try using it as a catalyst, hoping that it will be more effective than simple ammonium
chloride..
I'm not sure what you are implying with the alkoxide? There si no way any methanolate could be produced in the reaction with ammonia, presence of
water, no strong base, large amount of acetone. Or where you talking of something else?
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
Too tired to think it through at the present time, but it struck me as odd, we have an amine which forms a sodium salt... too fucking odd, there might
be something there but I'll have a look in the morning. Probably something to do with it acting as an acid, rather than a base, but the eyes are
shutting and its time for bed (In the CA they specifically referred to it as a salt).
The ammonium benzoate was used directly instead of ammonia and other ammonium salts, I think I pointed you toward the article once before? IIRC it was
suggested as a simple, lab-scale procedure for preparing small quantities of TAA. Sodium(?) benzoate isn't expensive, so I was thinking of grabbing
1kg of it, then forming the ammonium salt of it.
BTW Happy birthday Queen Elizabeth for yesterday, though why we celebrate it on Queen Victoria's birthday is fucking beyond me.
Whhhoooppps, that sure didn't work
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Well, you had mentionned the use of ammniumbenzoate, salicylate and anthralinate, but you couldn't find the article or something similar.
I might try the procedure with calcium zeolites, using ammonium nitrate and ammonium hydroxide, but I don't know where to find such zeolites. I will
have a look at aquarium stores or similar, and might try with silica gel or straight calcium chloride..
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Preparation of triacetoneamine according to US 6,646,127
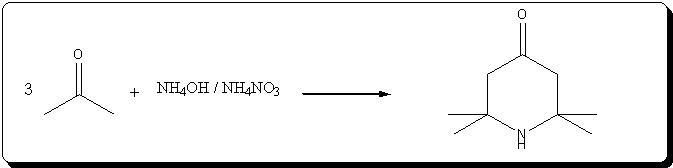
The patent's procedure in example 22 was followed, using CaCl2 as catalyst.
NH4NO3 was prepared by reacting 53% HNO3 with 30% NH4OH (very vigorous reaction), evaporating the water and drying the salt by melting it.
Acetone was technical grade, CaCl2 pellets were powdered prior to addition.
Procedure
In a 500mL 3-neck RBF, 5g (45mmol) of powdered CaCl2 and 40g ( 0.5 mol) of dry NH4NO3 wee suspended in 220mL acetone. 31mL (0.5 mol) of 30% NH4OH
(Panreac) were charged in the addition funnel, and slowly added dropwise, over 20min.


At first there was a little caking of the solids, then gradual dissolution. The flask warmed up to 40-45°C, so an ice bath was applied to keept temp
under 30°C. Once the addition was finished, most of the solids had dissolved to give a limpid solution.


The reaction medium was then stirred at RT for 7H total. The slightly yellow solution gradually turned orange then red, with that caracetistic
"fluorescent" red tint near the end.






The condenser was then replaced by a fractional distn setup, with a short (200mm vigreux column). The acetone was removed under slight vacuum at 45°C
over a 80°C oil bath. the reaction mixture had a strong smell of TAA. The colorless distillate smelled strongly of ammonia. Distn was continued
until no more acetone was distilled, and water started to take-off.n A dark red residu remained.


After cooling down to 30°C, the very viscous blood red residu whic smelled strongly of TAA was transfered to a 1L seperating funnel, and extarcted
with 4*50mL toluene. The extarcts came out golden orange, leaving the blood red color in the viscous aq. layer. After the 4 extraction, the aq. didn't
really smell as TAA anymore, but more as ammonia.


The toluene extracts were combined, washed with 50mL conc. K2CO3, and brine, thend ried over K2CO3 in a stoppered erlenmyer for 20min. The extract was
then transfered to a 500mL beaker, and acidified with 20% v/v H2SO4 in IPA. At first, a white precipitate formed, but as acidification continued, a
dark brown tar_like resin preicpitated also, very similar to the other TAA workups.

There was still a certain amount of white precipitate in suspension. After standing for 5min, most of the suspension was vacuum filtered, leaving
most of the resin in the beaker. 100mL acetone:IPA 50:50 were added, and the resin triturated, dissolving most of theresin and freeing more clear
solid. The suspension was vacuum filtered, thoroughly triturated on the buchner with 50mL acetone, and 50mL acetone:pet ether 50:50 before drying by
suction for 5min. The fine beige precipitate was then placed in a glass cup and dried under a lamp to afford 10.81g (41mmol) of crude
2,2,6,6-tetramethylpiperidone hydrosulfate.

The aqueous layer was re-extracted with 4*50mL AcOEt. These extracts took on a much redder colour. The combined extarcts were washed with 50mL co,nc.
K2CO3, brine, and dried over Na2SO4.


This extract was also acidified until acidic on dampened pH-paper with the same H2SO4/IPA, giving a very fien white precipitate at first, then a
brown/purple voluminous solid. This solid was much harder to correctly clean, giving a sticky paste, and required larger amounts of acetone, IPA and
AcOEt to get it relatively clean. The sticky paste finally dried, giving a crunchy brown solid (4.2g).

The two batches will be recrysatllied from MeOH/pet ether or MeOH/AcOEt.
Comments
Thsi reaction is pretty simple compared to the use of gaseous NH3, but the claime dyields are still not attained. Toluene seems to be a much more
selective solvent for the extarction than AcOEt is, giving a much cleaner product. The workup still needs optimizing though. Direct fractionnation
isn't possibble becasue of the large amounts of inorganic solids present in the syrup. Using diethyl ether as in the patent's procedure, or DCM wasn't
possible because of the ambient temp >35°C here (hot...). Tolueneseems pretty eefctive, but still requires large amounts of solvent. I guess a
gradaully distn of the solvent, and extarction with the recycled toluene could be the best option, as the extract seems pretty clean. Fractionnation
of the TAA freebase might be a better option than direct acidification, it is pretty clear there is a resinous product appering near the end of the
acidification, covering the TAA.HSO4.
Forming the hydrate or oxalate after fractionnation should give a pure product, stable as is. I will try another variation of the reaction, possibly
letting it heat up during the addition s is mentionned in the patent. I'm sure the yield is good, and that the isolation of the product is problematic
(TAA freebase is pretty soluble in water).
I think we are in the good direction though. Using only ammonia could be more practical, or at least catalytic amount s of ammonium nitrate (apart
from the extarction, producing dry NH4NO3 is the lengthy part).
I still have some NH4NO3 left, so will try another recation with a smaller NH4NO3/NH4OH ratio, but same acetone/ammonia donor ratio.
According to the patent, excellent selectivities to TAA are possible at RT, the acetone/ammonia ratio seems to be more important than the temp
regarding acetonine/DAA formation.
[Edited on 23-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
kmno4
International Hazard
Posts: 1503
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
I tried to bypass gaseous/liquid NH3 by mixing :
25g acetone
5g H2O
18g Ca(OH)2 , containing ~15% CaO - I just have very old CaO 
30g NH4Cl
Ammoniacates of CaCl2* are not very stable at room temp (at least higher than [Ca(NH3)2]Cl2), so I thought that Ca(OH)2/NH4Cl can serve as NH3 in situ
source. At the begining, mixture did not look good (semi-solid), almost not able to stirr. But after ~1 hour of shaking and stirring with glass rod,
consistency became more liquid than solid. It turn out to be stirable without problems with magnetic stirrer in a stopped flask. After ~24 hours of
stirring, mixture became white-red: red thick layer and semi-solid white reminder. There was some problem with removing this red something, but it
turn out (it was my 3rd run  ) that it can be removed with aid of dioxane. I just
added 4x20 ml of dioxane with stirring and careful pipetted off liquid after each portion of dioxane. ) that it can be removed with aid of dioxane. I just
added 4x20 ml of dioxane with stirring and careful pipetted off liquid after each portion of dioxane.
After evaporation there was ~20g of brown-red, very thick remainder, smelling amine (for me it was like morpholine smell), soluble in water, not
soluble in toluene, of very bitter taste. I supposed it was mixture of more than 10 substances but hoped there was large amount of acetonine in it.
So I treated it as acetonine and tried to convert into triacetonamine by procedure from some patent (acetonine, NH4Cl, KI, acetone, water).
Next, I evaporated acetone from water bath - I got brown-red sticky mass looking and smelling like input "acetonine"  . .
After adding 30% NaOH, formed oganic layer (red-brown) was taken, mixed with HCl(aq) and evaporated**. After very long evaporating from water bath, I
was not able to gain any crystals of hydrohloride. I got honey-like, brown-red mass.
That is all from me. Very messy job 
*if somebody is interested, I can share some articles about these ammoniacastes (U2U)
** propably I oversimplifyied this procedure - I should have extracted organic and aquenous layer with toluene (as Klute did)
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thank you for sahring! I am glad this subject is starting to interest others!
I have also tried a method using CaO/MgCO3, acetone , K2CO3 and NH4Cl, it turned the same as you describ it, a slurry turning to a paste under red
supernatant... It's been some time now (weeks?) an dI still haven't bothered doing the workup i admit. I will post a pic of it soon.
A efficiçent, practical preparation of TAA at a reasonable scale is still missing... I will try some more modification of the use of conc. aq.
ammonia, as this eems the most direct path. It's annoying becasue as long as this isn't solved, UI cannot do any furhter work on the nitrosyl
radicals and their modification. I would like to obtain a deacnt supply of TAA and work on from there, rather than preparing 5-10g each time..
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Ullmann
Hazard to Self
Posts: 51
Registered: 22-12-2004
Member Is Offline
Mood: No Mood
|
|
Very good work Klute and very inspiring and detailed procedures you are showing us, thank you!
I have a little something to add to the TEMPO field:
The reduced reactivity of the keto analog ask us to change that keto group in an OTC fashion and I think the best we can do is not to wolff-kishner it
nor reduce it but ketalize it!
check those few patents (no journal refs here, sorry)
EP1443049 & EP1457491 for ketalisation
EP1595868 & EP0574666 and US2005256312 for oxydation of the ketalized TEMPO
US5821374 and US5631366 for the oxydation of alcohols to aldehyde using ketalized TEMPO and TCCA or NaOCl
The most obvious choice and an example in the precited documents is the ethyleneglycol ketal. The yields with it are as good as with TEMPO and it is
the most straightforward to make in your garage! Only a few mol% are needed as catalyst for great and quick results! With a few g of it you can make
readily kg of aldehydes!
Be the TEMPO be with you!
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thanks alot
Indeed, the acetals weren't something I focused on, but do look promising. there aren't further ways of modifying them after, but they would
definatively be something to include in the oxydation test to come. I think I will get started again on the subject ins eptember or so, for now I'm
focusing on more time consuming projects as I'm on vacation. I would be very happy if you would like to join me and a few other in trying out various
derivatives, so then we could each report our results and see which are the most avantageous catalysts.. I plan on using the benzyl alcohol oxidation
asa comparasion test, because I have enough on hand, but other substartes could be used, like certain aliphatic alcohols.
Before any of this, a easy, high yielding method of preparing TAA would be needed to generate all the various derivatives, preferably withotu the need
of dry NH3 gas...
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
ziqquratu
Hazard to Others
Posts: 385
Registered: 15-11-2002
Member Is Offline
Mood: No Mood
|
|
I've just performed the synthesis of triacetonamine in accordance with Klute's post above and patent US 6,646,127. Due to the glassware I had
available, I did it on half of Klute's scale.
CaCl<sub>2</sub> (2.5 g) and NH<sub>4</sub>NO<sub>3</sub> (20 g) were stirred with technical acetone (110 mL) in a
3N, 250 mL RBF fitted with a reflux condenser, a thermometer and an addition funnel. Ammonium hydroxide (28 %, 15.5 mL) was added, with mild cooling,
over ca. 20 min, keeping the temperature below 20 °C. The cooling was removed and the mixture stirred at r.t. (20-25 °C) for 16 h. The originally
clear, colourless solution slowly became yellow, and was dark red by the end of the reaction.
The acetone was removed on a rotovap, and the resulting viscous aqueous layer was extracted with Et<sub>2</sub>O (5 x 60 mL). The combined
organics were dried over Na<sub>2</sub>SO<sub>4</sub>, filtered and the solvent removed to afford a yellow oil. The oil was
transferred to a smaller flask (it began to solidify in the process, so more ether was used to assist the transfer) and distilled through a short (~10
cm) Vigreux column to give the product ketoamine (15.60 g, b.p. ~80-87 °C/11 mmHg) as a yellow oil, which solidified to yellow needles on standing.
<sup>1</sup>H NMR (300 MHz, CDCl<sub>3</sub> 1.23 (s, 12,
CH<sub>3</sub>, 2.26 (s, 4 H, CH<sub>2</sub> 1.23 (s, 12,
CH<sub>3</sub>, 2.26 (s, 4 H, CH<sub>2</sub>
The distillation was somewhat bumpy - a bit of extra headspace would be an advantage here (in my case, the column JUST saved me when using a 50 mL RBF
- 100 mL would have been better). Also, following the extraction and removal of the ether, I ran an NMR, and the material was already quite clean -
just a little baseline rubbish. It would probably have been usable at that point without distillation. So perhaps ether is the best solvent for the
job (but, obviously, it isn't easily available to everyone).
I intend to perform the Wolff-Kishner reduction of the ketone in the near future. I'm unlikely to provide images, but I'll give a write-up if/when I
get to it!
|
|
|
mr.crow
National Hazard
Posts: 884
Registered: 9-9-2009
Location: Canada
Member Is Offline
Mood: 0xFF
|
|
Great work, and excellent thread. Now that it has been bumped to the top I say we sticky it
Has anyone preformed an oxidation using the 4-oxo-TEMPO? Is it necessary to remove the ketone group?
Double, double toil and trouble; Fire burn, and caldron bubble
|
|
|
KrysHalide
Harmless
Posts: 19
Registered: 29-11-2011
Location: France
Member Is Offline
Mood: No Mood
|
|
This review is of interest for anyone aiming the synthesis and derivation of 4-oxo-TEMPO:
C. Bruckner
Organic Preperations and Procedures Int 36 (); 1-31 (2004)
http://bruckner.chem.uconn.edu/PDFfiles/2004-OPP-oxoam.pdf
The H2O2/WO4 oxydation procedure was tried with succes:
In a 300ml erlenmeyer, 300mg of NaWO4.2H2O were dissolved in 50mL dH2O, followed by 4g of brown colored triacetoneamine crystals (>95%). A turbid
brown/orange solution was obtained.
3mL of 30% H2O2 were added with good stirring over 5 min, O2 bubbles appearing quickly.
The brown/orange solution was stirred two hours at RT, the colour fading to a limpid orange color.
The solution was saturated with K2CO3, a orange oil crashing out and quickly agglomerating to small orange crystals. These were extracted from the
milky aqueous with 3*25mL Et2O, the extracts washed with brine (with a spatula of NaHSO4 added), dried over K2CO3, and the ether removed on the
rotavap.
A clear dark orange oil was obtained. It was topped with hexane, and placed at -18°C. Orange cubic crystals were obtained, filtered, dried, to give
the product in a 87% yield, mp 35-36°C, pur by TLC (1/1 ether:toluene)
Never regret thy fall,
O Icarus of the fearless flight
For the greatest tragedy of them all
Is never to feel the burning light.
Oscar Wilde
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Nitric oxide is another stable radical with catalytic properties, though of course it has to be kept away from air.
For TEMPO, the wiki entry states that "The stability of this radical is attributed to the steric protection provided by the four methyl groups
adjacent to the nitroxyl group", but I do not think this is true.
Those methyl groups just prevent vicinal hydrogen atoms from ionizing off. An example is the oxidation of N,N-dialkyl-hydroxylamines by hydrogen
peroxide.
So I think N,N-di-tert-butyl nitric oxide would also be a stable radical. (tBu)2NO•
[Edited on 9-6-2013 by AndersHoveland]
|
|
|
KrysHalide
Harmless
Posts: 19
Registered: 29-11-2011
Location: France
Member Is Offline
Mood: No Mood
|
|
Ok, they made it before Klute could: a solid-supported TEMPO catalyst, with a simple as it get's preperation:
Silicagel, 4-hydroxy-TEMPO, and 1 eq of NaCl, RT then annealing..
Reuseable, easily recovered, efficient oxydation catalyst with no leaching.. Sounds promising and easily accesable.
"Novel [4-Hydroxy-TEMPO + NaCl]/SiO2 as a Reusable Catalyst for Aerobic Oxidation of Alcohols to Carbonyls"
N. Tamura, T. Aoyama, T. Takido, M. Kodomari, Synlett, 2012, 1397-1407.
DOI: 10.1055/s-0031-1290980
Let's hope other secondary oxydants can be used, although simply bubbling air looks like an economic alternative to traditional oxydants..
Could someone retreive the article? I've got 5g of 4-oxo-TEMPO waiting next to the NaBH4...
[Edited on 9-6-2013 by KrysHalide]
Never regret thy fall,
O Icarus of the fearless flight
For the greatest tragedy of them all
Is never to feel the burning light.
Oscar Wilde
|
|
|
KrysHalide
Harmless
Posts: 19
Registered: 29-11-2011
Location: France
Member Is Offline
Mood: No Mood
|
|
I have optimized the oxydation a bit, as I have noticed my first product was contaminated from unreacted TAA..
So the amine is dissolved in water, the tungsten salt added, and the 4eq of H2O2 added dropwise at RT. After an hour os stirring at RT, the solution
is gradually heated to 40°C (I once added the H2O2 at 40-50°C, there was an immediate color change but some resinous tar was also produced in small
quantities..).. After 2h at 40°C, NaCl is added until saturation, aswell as a spatula tip of NaHSO4 to keep Ph slightly acidic.. The mixture is
stirred vigorously as the NaCl dissolves, an red oil crashes out, letting the mixture to cool to RT. It is then placed the fridge for 30min: the oil
solidifies to a red disk. It is removed with a spatula, leaving an yellowish aqueous layer.
The red solid is dissolved in Et2O, diluted with an equal volume of hexane or any hydrocarbon (petroleum ether, ligroin, etc), dried with NaHSO4,
which absorbs a little brown color, decanted from the clumped salt, and the ether removed on the rotavap. The resulting red solution is placed in the
freezer, and turns to a thick paste of a pale orange solid, which filtered while cold, washed with a little cold hydrocarbon, and dried by suction.
The product is free from any unreacted amine.
The aqueous NaCl saturated layer is then stirred at RT, and more H2O2 added. Any unreacted TAA while have stayed in this aqueous layer, and gets
further oxydized on addition of further H2O2 as witnessed by the color change. I even added K2CO3 to this aqueous, and the white cristallin TAA
hydrate appeared. The biphasic mixture is stirred at 40°C for an hour or two, and treated as the first time. In this way, +95% yields of quasi-pur
4-oxo-TEMPO are obtained. Traces of degradation products are detected by TLC, but in minute amounts. Longer reaction times at RT could prevent this,
but it seems complete conversion of the amine at RT is VERY long..
My plan is to reduced the 4-oxo-TEMPO the the hydroxy compound, then esterify it with benzoyl chloride, or alkylate it. I will also try the
silica-supported catalyst is someone is kind enough to retreive the article I mentionned upthread!!
On the other hand, I will also go by another route, directly reducing the triacetoneamine with NaBH4, and then oxydizing the 4-hydroxy-TEMPO before
esterification/alkylation, to compare global yields. The oxydation might be smoother with the piperidol.
By the way, I tried sublimating a gram of 4-oxo-TEMPO, which was a complete failure, surely due to the fact that i did it at atm pressure.. The
red/orangfe solid was slowly heated in a RBF with a cold finger, but only a liquid vapour was deposited, with evident decomposition (turned to tar,
generation of water vapour), and these drops never solidified. I was scared that this would happen with vacuum applied, considering the very low mp of
these TEMPO derivatives..
I am not familiar with sublimations, is vacuum indispensable? Or do I have to heat less, and leave it to it own device for several hours?
Never regret thy fall,
O Icarus of the fearless flight
For the greatest tragedy of them all
Is never to feel the burning light.
Oscar Wilde
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
Quote: Originally posted by KrysHalide  | I have optimized the oxydation a bit, as I have noticed my first product was contaminated from unreacted TAA..
I am not familiar with sublimations, is vacuum indispensable? Or do I have to heat less, and leave it to it own device for several hours?
|
For most compounds, a high vacuum is indispensable. Otherwise the compound merely oxidizes from oxygen in the air or decomposes from the heat before
it sublimes. The lower the vacuum, the easier most compounds will sublime at lower temps.
Unfortunately, I can't get access to that article right now, but it looks interesting. There are several companies that sell other oxidants
covalently bound to resins, but this looks very simple, inexpensive, and easy to use. Very nice find.
[Edited on 12-6-2013 by Dr.Bob]
|
|
|
KrysHalide
Harmless
Posts: 19
Registered: 29-11-2011
Location: France
Member Is Offline
Mood: No Mood
|
|
okay, thanks for the explaination! But, as vacuum decreases the bp, will it not also decrease the mp? Meaning that under reduced pressure, 4-oxo-TEMP
will stay as a liquid even at -10°c (cold finger temp)? Or is the mp not affected?
Never regret thy fall,
O Icarus of the fearless flight
For the greatest tragedy of them all
Is never to feel the burning light.
Oscar Wilde
|
|
|
watson.fawkes
International Hazard
Posts: 2793
Registered: 16-8-2008
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by KrysHalide | | okay, thanks for the explaination! But, as vacuum decreases the bp, will it not also decrease the mp? Meaning that under reduced pressure, 4-oxo-TEMP
will stay as a liquid even at -10°c (cold finger temp)? Or is the mp not affected? |
Melting points aren't
much different between atmosphere pressure and vacuum, since the absolute pressure difference isn't much as pressure differences go. Nor does it
change the partial pressure of the material very much. What it does is to change the transport dynamics for sublimation. When there's ordinary
pressure at a surface, any molecule that comes off the surface won't travel very far (see "mean free path") before colliding with some other gas
molecule . There's a deposition-sublimation equilibrium that gets set up. Some molecules diffuse away, but that's a slow process. This is called the
"viscous flow" region. You have to wait for them to eventually diffuse to the other side.
With vacuum, the mean free path increases. If that length increases to be larger than the size of your geometry, you move into what's called the
"ballistic flow" region, where there are very few collisions. In a sublimation apparatus, molecules sublime off the hot surface (in random
directions), fly through vacuum with no collisions, and deposit on the cold surface. If you try this without vacuum, you're in the viscous flow
region. Transfer rates can be orders of magnitude lower. For some substances that readily sublime, this may be adequate.
As a rule, you want to run the hot surface as hot as you can without either decomposition or overwhelming your cooling system, you want to run the
cold surface at just under the deposition temperature (to minimize radiative heat transfer), and you want to ensure that you're vacuum is low enough
that you're in the ballistic flow region for the gap between the two. Higher vacuum beneath that threshold is mostly wasted.
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
Requested by Klute
Novel [4-Hydroxy-TEMPO + NaCl]/SiO2 as a Reusable Catalyst for Aerobic Oxidation of Alcohols to Carbonyls
N. Tamura, T. Aoyama
Synlett
2012; 23(9): 1397-1401
DOI: 10.1055/s-0031-1290980
Attachment: phpGGs8p7 (207kB)
This file has been downloaded 1401 times
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
Thanks Solo, that is an interesting paper, and gives a pretty simple way to prepare a useful oxidant with very simple chemicals. I have used some
similar solid supported reagents, and they are very handy for some uses, where having to purify something from the oxidants would be a pain. I may
have to try that sometime for making aldehydes.
|
|
|
KrysHalide
Harmless
Posts: 19
Registered: 29-11-2011
Location: France
Member Is Offline
Mood: No Mood
|
|
Yes, thank you very much Solo!!
I have good news: I will be able to try this procedure very soon, as my first bath of 4-hydroxy-TEMPO is drying right next to the laptop!!
Reduction was very smooth, and gave a very clean product in quantitative yields!!
Here's what I did:
3.2g of 4-oxo-TEMPO were dissolved in 30mL abs EtOH. The dark red solution was cooled in a ice water bath, and 0.55g of NaBH4 were added over 5 min.
there was slight H2 evolution, and a exothermic reaction. The ice bath was removed, and the reaction medium stirred at RT for 3H. There was a subtil
color change, from dark red to a more orangish red.
Most of the ethanol was then removed on the rotavap at atmospheric pressure (vacuum pump down), and 20mL of water were added, along with some solid
K2CO3 to attempt crashing the product out as during the TAA oxydations. But there was too much EtOH left, and a orange organic layer seperated. The aq
was diluted with a little more water, and the product extracted with ether, which did the job very well. The combined extracts were washed with 2*25mL
saturated K2CO3, and dried over solid K2CO3. The ther was then removed at atm. Hexane was added to the residu, but this time two layers formed, the
product oiled out unlike 4-oxo-TEMPO. A little Et2O was added to try and obtain a homogenous solution, and the flask placed in the freezer. No
cristallization occured even after 1H, so the flask connected to the rotavap, and as much solvent was removed as possible. Petroleum ether was added
to the residu, and while cooling the flask under a stream of cold water, the product solidified nearly instantly, giving a pale orange solid, looking
alot like 4-oxo-TEMPO, but of a much lighter pale orange color. The flask was left in the freezer for 30min to insure complete solidification, and the
product vacuum filtered. The chunks of solid were crushed as fine as possible, triturated with some petroleum ether, and filtered again. After air
drying, 3.2g of a light cristalline pale orange powder were obtained, surely still a little solvant present.
TLC ( Et2O) reveals no 4-oxo-TEMPO, and very faint impurities spots in addition to the product.
Succes
Tomorow, part of the product will be reacted with benzoyl chloride, and a small scale experiment will be conducted, the oxydation of benzylalcohol
with 4-hydroxy-TEMPO. I haven't decided on the conditions I will use, I have TCCA, Ca(OCl)2, NaBrO3 and a few other secondary oxydants I could use..
I guess i will go along the sides of substrate dissolved in DCM, catalyst added, and solid oxydant graddually added, in presence of TBAB if need be. I
will have to check the different procedures published, haven't read them in while.
I will also try the SiO2 supported version, but with a slight modification of the published procedure in that I will not use atmospheric oxygen as
sec. oxydants. I guess this will circonvent the need for NaCl additive.
Any comments, suggestions, welcomed.
Never regret thy fall,
O Icarus of the fearless flight
For the greatest tragedy of them all
Is never to feel the burning light.
Oscar Wilde
|
|
|
KrysHalide
Harmless
Posts: 19
Registered: 29-11-2011
Location: France
Member Is Offline
Mood: No Mood
|
|
Ok, the oxydation proceeded well.
1.52g (10mmol) of 3,4-methylenedioxybenzyl alcohol were diluted in 25mL of DCM, 50mg of 4-hydroxy-TEMPO were added. Then a solution of 520mg of NaHCO3
and 120mg of KBr and 80mg of TBAB in 20mL H2O was added. The biphasic mixture was cooled to 0°C in a ice bath, and 7mL of 10.2% NaOCl solution was
slowly dripped in with good stirring. The first drops caused a quick milkyness, and appearance of a yellow color in the aq, which dissipated after a
few seconds. Surely formation of the oxonium salt, and it's disappearance after reaction with the alcohol? The addition rate was adjusted to let time
for each drop to correctly react, and took 30min. The bottom organic layer displayed a yellow/green color, and a white emulsion/fine solid appeared
gradually. The reaction was followed by TLC, and was slower than expected. The benzylic alcohol was present even after 1H reaction time, so 3 more mL
of the NaOCl solution was slowly added.
TLC reavealed formation of two compounds, with similar Rf, one darkening after exposure to UV lamp. A small uneluted spot also appeared near the end
(carboxylic acid?). The aldehyde spot beacme larger with room temp stirring.
After 2H30 total reaction time, the biphasic mixture was filtered on a N°4 glass fritt to remove the white emulsion "blobs", the organic seperated
and washed with NaHCO3 solution two times, and was then dried over Na2SO4. It was then filtered over a 2cm plug of silicagel, to remove the catalyst,
which was quite effective by the color of the silicagel. The plug was rinced with DCM, and the solvent removed, to afford a green oil, that
crystallized on cooling in the fridge. The mp was depressed, surely beacause of minor impurities. A 74% yield of crude product was obtained.
The aldehyde was directly used in a aldol condensation with acetone catalyzed by NaOH, which is currently stirring.
Basicly, this works well, but I was surprised by the slow reaction rate after reading numerous articles claiming that the reaction is over in minutes.
The reaction conditions will be optimized, using different secondary oxydants, and TEMPO derivatives.
I also obtained some 2,2,6,6-tetramethylpiperidine, that I will oxydize to TEMPO, so I can compare the efficienty of each derivative to the original
catalyst.
Unfortunaly, it will be on another substrate, as I only have 500mg of piperonyl alcohol left.
[Edited on 14-6-2013 by KrysHalide]
Never regret thy fall,
O Icarus of the fearless flight
For the greatest tragedy of them all
Is never to feel the burning light.
Oscar Wilde
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
So, I decided to have a pop at this myself, following US patent 3959295. This is the same patent that Klute made use of at some point in this thread,
although I cannot find mention of using CaCl2 in the patent. I'll spare the details on the preparation, but at the end of the reaction (I used
judgement based on what the patent says) I filtered off the ammonium chloride (it never went into solution, another difference to Klute's replication)
and concentrated to a residue. On standing the dark red-brown residue crystallised to a semisolid. I figured I'd bypass the issues with precipitating
a solid, and directly vacuum distilled the semicrystalline residue at ca. 7mbar. Three fractions were taken, over the range 64-84*C and 6-9mbar. All
three fractions were yellow oils, and the third crystallised on standing. Small samples were dissolved in methanol and TLC'd in 50%v/v EtOAc/n-Hexane,
and visualised with UV, Ninhydrin and 2,4-DNPH (see attached image). The spots are a little strong, but what I suspect is the product (mostly fraction
3) does not stain particularly well with ninhydrin. I was actually suprised that there were so many impurities.
Where to go from here? Well I guess theres the option to purify further by hydrate or salt formation. I've not really decided what to do yet though,
so any suggestions/comments are welcome. Its worth noting that Ziquarratu (sp?) also went the route of distilling the product directly, although he
did not comment on the purity of his distillate.

|
|
|
ziqquratu
Hazard to Others
Posts: 385
Registered: 15-11-2002
Member Is Offline
Mood: No Mood
|
|
It was quite some time ago, and I've moved to a new work-place and thus don't have the data or the sample any more. However, from memory, the purity
following distillation was excellent (and I suspect the slight amount of garbage - invisible by NMR, but a small high-running spot by TLC) came from
my poor choice of distillation apparatus (too small with lots of bumping).
One thing I do recall, however, is that the compound was quite unstable - stored in an air-tight bottle flushed with argon, it darkened quite quickly,
becoming black (but still crystalline, for the most part) within a month or so. Could have been due to impurities, or perhaps light was the problem -
I never did investigate as I ended up not needing the material.
|
|
|
| Pages:
1
2
3
4
5 |
|









