| Pages:
1
..
23
24
25 |
Etanol
Hazard to Others

Posts: 218
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
The experiment showed that an excess of ascorbic acid at pH <3.5 reduces Cu(++) in solution to a metallic copper in a few hours at room temperature
or in 3-5 minutes in a hot solution.
At PH>3.5, Cu(OH)2 is formed, that is instantly reduced with ascorbic acid to copper (I) oxide Cu2O at room temperature.
How to avoid these impurities in DBX-1 synthesis?
[Edited on 15-8-2024 by Etanol]
|
|
|
Niklas
Hazard to Self
Posts: 81
Registered: 1-12-2023
Location: Germany
Member Is Offline
Mood: Polymerized
|
|
Synthesis of 1-Diazidocarbamoyl-5-azidotetrazole (C2N14)
So, for quite a while now I‘ve been wanting to try making some 1-diazidocarbamoyl-5-azidotetrazole, commonly referred to as “azidoazide azide“,
as it’s one of the more chemically interesting energetics in my opinion, and it has quite a reputation of being the most sensitive one out there.
While the second claim has pretty much been disproven by YouTubers like ReactiveChem and Explosions&Fire [1][2], for being completely heavy-metal
free I still consider it to be quite an impressive substance, and kinda wanted to see for myself.
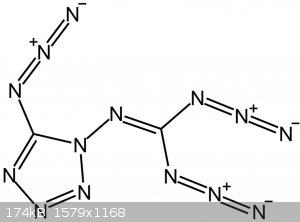
Fig.1: Chemical structure of C2N14
I‘ve started this project around two years ago with the synthesis of disodium azobistetrazolate pentahydrate, but I never really came around to
continuing since then, so the salt had been sitting in storage ever since. This finally changed last week, and I managed to successfully make the
desired tetrazole, as detailed below.
The 1961 approach by Grundman and Joseph [3]:
This is the approach most generally employed, and the synthetic details are given in Engager’s publication, which was my primary source for the
entirety of the preparation [4].
The synthesis starts from 5-aminotetrazol (5-ATZ), which gets oxidatively coupled to the above mentioned azobistetrazolate, which then serves as a
precursor to the isocyanogen-system by reaction with elemental bromine. The resulting tetrabromide can then undergo simple substitution with azide,
and the resulting “open form“ of C2N14 (isocyanogen tetraazide) quickly cyclizes resulting in the desired end product.
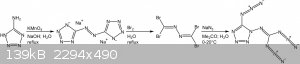
Fig.2: The 1961 synthesis of C2N14
Disodium azobistetrazolate pentahydrate:
As I did this synthesis quite a while back, and my labnotes were rather scattered at the time, I unfortunately don’t have any of my experimental
details on hand, though in the end I just followed the procedure given in [4] pretty much exactly, only adding the potassium permanganate as a
saturated solution rather than as a solid, so by checking on it you should still be able to follow.
As one may expect from the nitrogen-rich structure this intermediate is already an energetic itself, and while testing some crystals of it on some
aluminum foil does cause some rather significant damage (footage of some of its detonations can be found in Extractions&Ire‘s video covering
this preparation [5]), as it’s a pentahydrate salt it’s impossible to properly confine it though. While it would therefore generally be advisable
to store it wet, but as I didn’t notice any shock or friction sensitivity it was simply kept as the dry crystals shown.
 
Fig.3-4: Synthesis of disodium azobistetrazolate and carefully evaporating down the final solution
 
Fig.5-6: The product under the microscope

Fig.7: Crystals of disodium azobistetrazolate pentahydrate
Isocyanogen tetrabromide:
A 100 ml three neck round-bottom-flask, equipped with a Dimroth-condenser, was charged with 0,75 g of elemental bromine (9,4 mmol; made by the
oxidation of sodium bromide with potassium persulfate in 95,6% yield), suspended in 20 ml of dest. water. To this, while stirring, a solution of 352,3
mg of disodium azobistetrazolate pentahydrate in 15 ml dest. water was slowly added dropwise, at which point some gas evolution quickly became
evident. The mixture was refluxed in a heating block for 3 h, during which time the red bromine color increasingly discolored, and a red oil
separated, which eventually “steam-distilled“ into the condenser collecting as a beige solid. The condenser was washed out with DCM, the light
yellow reaction mixture extracted more three times, and the pooled extracts washed with saturated brine and dried over anhydrous sodium sulfate. The
solvent was removed by rotary evaporation, resulting in a yellowish-green oil, which solidified into a crystalline solid on cooling to room
temperature. This crude product is sufficiently pure to be used in the final step of the preparation. 136 mg, 31,3%
 
Fig.8-9: Synthesis of isocyanogen tetrabromide

Fig.10: the collected crystalline isocyanogen tetrabromide
1-Diazidocarbamoyl-5-azidotetrazole:
In a 10 ml falcon-tube 25 mg of the isocyanogen tetrabromide (67,3 μmol) were dissolved in 0,3 ml of acetone and and the greenish solution cooled to
around 0°C with the help of an icebath. The mixture was purged with argon for around a minute, and the tube loosely capped to ensure somewhat inert
if an atmosphere. To this 150 μl of a solution of 0,9 g sodium azide in 6 ml dest. water (346,1 μmol), cooled in the same icebath, were slowly added
dropwise with occasional swirling of the reaction vessel, and things were then left to stand first for 1 h at 0°C and then 3 h at room temperature.
After this 2 ml of dest. water were added to the now orangish solution, and things left to stand in the icebath for 45 min to allow the product to
separate. Nothing really happened though until the walls of the tube were very carefully scraped with a plastic pipette, on which a white crystalline
solid gradually precipitated. This was removed by gravity filtration, the residue washed with some more water, and the wet white crystals used as such
for further experimentation and energetics testing. The footage of those tests can be found in [6].

Fig.11: Synthesis of C2N14
 
Fig.12-13: The reaction mixture before and after the addition of the water
The 2011 approach by Klapötke [7]:
This approach seems much less accessible on first glance, starting from triaminoguanidine hydrochloride in the original publication. This precursor
can actually easily be prepared from widely available guanidine hydrochloride though, and I personally based myself on the procedure given in [8].
This doesn’t just make it a particularly easy approach though, and the key-dimerization (for mechanistic details check out [7]) actually is really
pH-sensitive to the extent that tracking the pH with pH-paper doesn’t really suffice [9]. Therefore I may revisit things as soon as I get a proper
pH-probe.
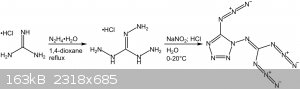
Fig.14: The 2011 synthesis of C2N14
Triaminoguanidine hydrochloride:
In a 50 ml round-bottom flask, equipped with a Liebig-condenser, 970 mg of guanidine hydrochloride (10 mmol) were dissolved in 25 ml of warm dioxane
and 3,7 ml 100% hydrazine hydrate (75 mmol) quickly added over the course of one minute. The colorless mixture was heated to a reflux in a heating
block for 3 h, with some ammonia generation quickly becoming evident (detectable by holding some wet pH-paper over the opening of the condenser), and
a white solid starting to precipitate. Things were cooled left to cool down over night, during which time the mixture seemed to have pulled some
moisture from the air, so around 10 ml of the solvent were quickly removed by heating on the hotplate, and the precipitated solids removed by vacuum
filtration after things had cooled back down again. The residue was washed with some diethylether and acetone, and the very slightly pinkish* crystals
dried on the air. 983 mg, 70%
*This discoloration later turned a slight yellow, it can generally be expected to get a completely white product though as this presumably just came
from the glassware being contaminated.
 
Fig.15-16: Synthesis of triaminoguanidine hydrochloride

Fig.17: The collected slightly pinkish crystals of triaminoguanidine hydrochloride
1-Diazidocarbamoyl-5-azidotetrazole (failed attempt):
In a 50 ml falcon-tube 70,5 mg if the triaminoguanidine hydrochloride (0,5 mmol) were dissolved in 7,5 ml of dest. water, and 1 ml of 1 molar
hydrochloric acid (1 mmol) was added to the colorless solution. Things were cooled to around 0°C in an icebath, a solution of 69,5 mg of sodium
nitrite (1 mmol) in 7,5 ml dest. water was then very slowly added through an addition funnel while magnetically stirring, and the stirring continued
for 45 minutes at room temperature afterwards. The pH was very slowly brought to exactly 8* by dropwise addition of 0,1 molar sodium hydroxide
solution, during which the previously colorless solution gradually turned a yellow color. Things were extracted three times using 15 ml of
diethylether each, by shaking inside the falcon-tube and removing the top layer with the help of a pipette, and the yellow extracts passed through a
silica-filled syringe in hopes of removing some amine impurities which would otherwise require the use of a short chloroform column after isolation.
The now light yellow extracts were carefully evaporated down in a 50°C water bath while, on which only a tiny quantity of a totally non-energetic
yellow oil remained though, why this attempt was discarded.
*This was achieved using pH-paper in this run, what, as mentioned above, isn’t sufficiently precise for this reaction, why this presumably failed.
 
Fig.18-19: Failed attempt of synthesizing C2N14
 
Fig.20-21: The mixture before and after the addition of the base
Sources:
[1] https://youtu.be/Hv4Ulb16imk?si=9fL8qJpg2KFjD6S2
[2] https://youtu.be/-Sz4d7RQB6Y?si=Fcq-Es_7Y0mnt1rh
[3] https://patents.google.com/patent/US2990412A/en
[4] https://sciencemadness.org/scipics/Energetic_Derivatives_Of_...
[5] https://youtu.be/_m52RquJi_M?si=7KKYR0Xm8ZWyvvWr
[6] https://youtu.be/Ma-D-cARvXw?si=fsCMM69PBkL09AuV
[7] https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.2011003...
[8] https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002...
[9] https://youtu.be/zUdBHx-F8wQ?si=OXGysXIyVCp9Loo7
|
|
|
BAV Chem
Harmless
Posts: 29
Registered: 9-5-2021
Location: Irgendwo im nirgendwo
Member Is Offline
Mood: Thoroughly confused
|
|
Wow, amazing work Niklas!
This might be one of the nicest looking documentations of this synthesis.
Quote: Originally posted by Niklas  |
The 2011 approach by Klapötke [7]:
This approach seems much less accessible on first glance, starting from triaminoguanidine hydrochloride in the original publication. This precursor
can actually easily be prepared from widely available guanidine hydrochloride though, and I personally based myself on the procedure given in [8].
This doesn’t just make it a particularly easy approach though, and the key-dimerization (for mechanistic details check out [7]) actually is really
pH-sensitive to the extent that tracking the pH with pH-paper doesn’t really suffice [9]. Therefore I may revisit things as soon as I get a proper
pH-probe. |
To my knowledge there's one other person on the internet, that being ReactiveChem on Youtube, who tried this and he got the same results, probably
because of bad temperature control.
I myself actually also tried this exact reaction about a week ago, albeit with lower quality reagents, and it actually worked on the second attempt so
here are my two cents on that:
Triaminoguanidinium nitrate was prepared by refluxing 2,44g of guanidine nitrate (20mmol), 9,2g of hydrazine sulfate (70mmol) and
5,7g of sodium hydroxide (140mmol) in 10ml of water for 5h (adapted from [1]). Afterwards while still hot 25ml of cold water with a few drops of conc.
HNO3 were added through the condenser. (This serves to acidify the reaction mixture and prevent reactions of free base aminoguanidines with oxygen in
the air. This is also likely what caused the pink coloration in your run as oxidation leads to the formation of tetrazine derivatives [2] which are a
deep red/purple.) Over the course of a day a bunch of product crystallized at room temperature and more was recovered by fractional crystallization.
Yield was 1,7g of of TAG nitrate (67%) which was recrystallized before use.
Now the fun part, C2N14:
For the diazotization 0,33g of triaminoguanidine nitrate (2mmol) and 0,7g of HCl (22-25%, I don't know the actual conc., hardware store grade) were
dissolved in 30ml of water and cooled in the freezer. 0,62g or 4mmol of 45% NaNO2 (homemade, the rest is nitrate) was dissolved in 30ml of water and
cooled in an ice bath. Once the TAG/HCl solution was semi-frozen the nitrite was added to it with stirring over the course of 20 minutes. During this
step the solution was kept in a partially frozen state by occasionally cooling it in an ice/salt bath @-18°C. This makes temperature control way
easier. Residual ice was then allowed to melt and after that point stirring was continued for another 30 min. Then 0,1g of ammonium chloride was added
and allowed to react for another 10 min. (The idea here is to remove residual nitrite. I figured it can't hurt, no idea if it actually helped though.)
The solution was basified by slowly pouring in 0,1M NaOH solution until the pH was a little above 8 (determined using cheap, chinese pH paper).
Interestingly during the dimerization/cyclization step I found the pH to decrease again over time so I kept adjusting it to 8 for 5-10 minutes. Then
the solution was extraced with three 50ml portions of ether. Evaporation yielded a few tiny droplets of a yellow oil which was highly explosive,
detonating even in really small amounts so I'm rather certain it was mostly C2N14. Most likely purer reagents would actually give a crystallizable
product or perhaps there's really no way around column chromatography . .
That was my second go at it. On the first run I didn't freeze the solution, didn't add any NH4Cl, let the pH drift back without realizing and used
ether that wasn't stored over KOH (might've contained some acid). Klapötke et al. wrote that residual nitrite will destroy the azide groups if the pH
is too low [3] so that's why i added the ammonium chloride. This might also mean that bringing up the pH too carefully and too slowly could actually
degrade the product.
[1] US5041661
[2] The chemistry of aminoguanidine and related substances
[3] C2N14: An Energetic and Highly Sensitive Binary Azidotetrazole
|
|
|
Niklas
Hazard to Self
Posts: 81
Registered: 1-12-2023
Location: Germany
Member Is Offline
Mood: Polymerized
|
|
Thank you!
Good to know it’s likely just the temperature causing issues, while I didn’t notice any microdetonations like ReactiveChem, there was still
definitely some gas generation (as can be seen on the pictures), so I likely just wasn’t careful enough with the nitrite addition. Well it may be
both factors causing issues, but I do think adding ammonium chloride to destroy excess nitrite does indeed help with the pH-sensitivity, so it’s
something I‘ll definitely give a shot as well when revisiting things.
|
|
|
Microtek
National Hazard
Posts: 920
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
In my search for an effective primary that poses less of a health hazard, I came across references to nitriminotetrazolates, both here on SM and in a
number of papers. I tried a number of different methods of synthesis, such as dehydration of 5-ATz*NO3 with sulfuric acid, nitration of 5-ATz with
anhydrous HNO3 and mixed acid nitration of 5-ATz. I found that the latter worked the best for me.
The method I used is as follows:
- 5.3 g anhydrous 5-ATz (from drying the hydrate at 110 C) was dissolved in 12.5 ml conc. H2SO4 in portions with stirring, keeping temp below 40 C
using a cooling bath. The solution was somewhat opague when all of the 5-ATz had been added.
- Ice was added to the cooling bath and when temp had reached 20 C, 5 ml WFNA was added dropwise, keeping temp in the 20-30 C range. The soln cleared
up as the nitric acid was added.
- When nitric acid addition was complete the cooling bath was removed, and stirring was continued for 15 minutes at room temp.
- The soln was poured on about 35-40 g of crushed ice, and NH3(aq), 25% was added to a pH of 5-6 with cooling keeping temp below 35 C. I used pH
paper, but there is a subtle colour change when the pH approaches neutral.
- Towards the end of addition of ammonia, a white precipitate had begun to form, and on cooling to 5 C in the refridgerator, a large amount of product
had settled out. This was collected by suction filtration and was with ethanol in the Büchner filter (I should probably have washed with a little
cold water first to remove the last traces of AN and ammonium (hydrogen) sulfate).
On drying, I had collected more than 7 grams of product which is about 90 % yield depending on the degree of deprotonation of the nitriminotetrazole
(it could precipitate as either mono- or diammoniumnitriminotetrazolate).
To verify the identity of the product, I combined a solution of it with AgNO3 which gave a white precipitate that behaved much like AgNT. With
Pb(NO3)2 it made a white precipitate that was energetic, but did not DDT in small amounts at first. However, when dehydrated in the oven at 150 C, it
came to resemble LA in vehemence and power.
I also tried with Fe(II) and Fe(III), but neither of those gave much. On the other hand CuSO4 soln turned deep green and over the course of a few
minutes gave an about quantitative precipitate of malachite green precipitate. On washing and drying it was at first not very impressive, but when
dried in the oven turned a drab olive and became as powerful and vehemic as the silver salt. Note that upon oven drying a second (very small, less
than one mmol) batch at 200 C, it detonated in the oven and shredded the paper weighing boat it was in.
I am aiming for Ca-NITz, the calcium salt which according to Klapötke is the most promising LA replacement (and it should by almost as non-toxic as
iron salts), however it has so far eluded me.My (NH4)xNITz does not produce an energetic precipitate with CaCl2, neither does it work if I react the
(NH4)xNITz with NaOH to make sure I have deprotonated it fully.
My next attempt was to isolate the free acid (which according to Klapötke has a calculated shelf life of 58 years) by reacting an aqueous soln of 0.5
g (NH4)xNITz with sulfuric acid and extracting with successive 5ml portions of DCM. The two first portions gave a fair amount of solid when
evaporated, but the third was negligible, so I think DCM is a good choice of solvent for the extraction.
I intend to react a soln of H2NITz with solid CaCO3 or Ca(OH)2, but haven't gotten that far yet.
|
|
|
dettoo456
Hazard to Others
Posts: 255
Registered: 12-9-2021
Member Is Offline
|
|
Was the hygroscopicity/water complexation mentioned by Klapotke with the Ca salt? I don’t recall whether it was the 5-NTz or 5-NITz Calcium salt
that was talked about before on an SM thread, but I do remember the author speaking about its tendency to revert to a puddle in ambient conditions.
Anyways, you probably found this thread already, but here’s Hey Buddy’s notes from 2023:
“ Metal Nitrimino Tetrazoles continued...
Ba, Sr, Ag all displaced successfully, Ca is inert, which seems like it has some error in preparation, as CaNATz should be explosive.
The other three salts were dried gently and probed for flame responsive detonation. Ba and Sr gave micro popping on burning.
Ba pops were the strongest but there was no observed detonation.
Ag deflagrates.
These salts as tested, should all be in their hydrated forms with whatever crystal water they have. Presumably if heated, they would become anhydrous
which would make them more sensitive and easier to achieve detonation. The Barium and Calcium salt are both known to have different performances and
sensitivities in hydrated form versus anhydrous.”
|
|
|
Axt
National Hazard
Posts: 861
Registered: 28-1-2003
Member Is Offline
Mood: No Mood
|
|
I played with nitriminotetrazolate on page 3, made through nitrosation of aminonitroguanidine and found the potassium salt feeble and the silver salt
to "snapple", i.e. individual crystals would snap and spread the rest of the mass around without propagation, have you witnessed that Microtek? At
least as a response to unconfined flame, this may be different under confinement or indeed may have remained bonded water. Did you derive any product
at all from the aminotetrazole nitrate + H2SO4 attempt?
Back then I made the assertion that literature mentioned they had no initiating properties, so I've had a look for where I derived that from. I
believe it's from the attached DoD report. It seems they were interested in stab sensitivity and all tested were very insensitive (Ag salt 22cm drop
of 2.5kg weight). They attempted and failed dehydration of the copper salt, but it was only heated up to 50C in vacuum.
In other related news, silver azotetrazolate, very vehemic, on par with silver nitrotetrazole but significantly more sensitive (26-32mm drop of
1.025kg weight, silver nitrotetrazolate required a 56mm drop). It's an awful brown volumous precipitate that shrinks 1000 fold on drying into caked
dark brown plates. The copper salt was very fine and mostly passed through the filter paper, the small bit that was caught had a slight tendency to
"snapple" but DDT'd readily on ignition.
Attachment: Metal Salts of Nitraminotetrazole.pdf (1.1MB)
This file has been downloaded 47 times
|
|
|
Microtek
National Hazard
Posts: 920
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
Yes I did note that "snapple" behaviour with the silver salt. Under Al foil confinement it detonates very powerfully. The 5-ATz*NO3 + sulfuric did
give some product that did in fact give an explosive precipitate with AgNO3, so I'm sure it does work. Just not as well as the one I described above,
in my experience (which I found in a patent on gas generants for airbags). I'm sure it would be possible to tinker with the reaction to get a decent
yield using the other method, and it would be convenient to avoid having to distil HNO3.
I used some of the free acid to prepare the calcium salt by adding the calculated amount of CaCO3 to a soln of H2NITz in water. The CaCO3 dissolved
with liberation of CO2 but did not immediately precipitate anything. As I added the last of the CaCO3, solid matter could be seen suspended in the
soln, but it is unclear if this was precipitated product or unreacted CaCO3. The water was evaporated with moderate heat (hotplate setting of 130 C)
in an airstream from an aquarium pump, and the white solid residue was tested. It was found to be explosive, but only weakly so. I then dried it in
the oven at 200 C (CaNITz is stable up to 360 C, according to Klapötke, et al) and recorded the weight loss. after 30 min, no further reduction in
mass could be observed and on testing, the product was much more vehemic in character though it still fell a little short of my expectations.
It was then discovered this morning that blocky crystals had separated from the filtrate. On quite a lot of heating these detonated with great
violence. I need to do more structured experiments with extended duration precipitation, but this has given my hope that CaNITz may be able to fill
this particular niche.
Edit: It seems I made a mistake about DCM being a suitable extraction solvent, at least on its own. What I actually used was a mixed DCM/THF solvent,
and pure DCM doesn't work at all. On the other hand, I discovered that ethyl acetate works well, although it may hydrolyse slightly from contact with
the quite acidic H2NITz.
Also, the blocky crystals were dried in the oven at 150 C for 30 minutes. This led to a marked change in appearance from translucent glittering
crystals to a white somewhat amorphous solid. This was accompanied by a ca. 40 % mass loss which corresponds quite well with a CaNITz*5H2O -->
CaNITz transition.
The dried solid is very powerful and very vehemic as well, detonating without confinement both when cooked off with flame beneath Al foil, and also on
flame contact from above. I measured out 0.010 g and divided it into 4 approximately equal piles, then subjected one of these piles (2-3 mg) to a
"flame from above" test. It detonated with a very sharp report and perforated the foil.
[Edited on 17-2-2025 by Microtek]
|
|
|
Microtek
National Hazard
Posts: 920
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I did another experiment where H2NITz was reacted with CaCO3 until neutral. The solution was filtered to remove the small amount of unreacted CaCO3
and the mix was heated to reflux for 2 hours. It was then cooled to 5 C in the fridge overnight. The next morning a decent yield of the blocky
crystals had formed and was filtered off, washed with cold water and then ethanol. On drying they perform identically to the other batch. It seems
that this reaction works a lot better if the mix is heated before setting it aside for precipitation.
I then considered the way Klapötkes group prepares CaNITz (they nitrate 5-ATz with 100% HNO3 and then neutralize the diluted mix directly with
Ca(OH)2), and thought that would be equivalent to dissolving NH4NITz in dilute acid (HNO3 or HCl) and then neutralizing with CaCO3 or Ca(OH)2. I
therefore dissolved about 10 mmol NH4NITz in 40 mmol dilute nitric acid and added 20 mmol CaCO3 in small portions. Before the end of addition,
substantial amounts of white precipitate had formed, so I stopped addition when no more gas evolution could be detected. pH was still around 2-3 at
this point. On cooling, the mix had become almost solid with precipitate, so I diluted with water, filtered and dried.
It turned out to be only weakly energetic and after a lot of bug hunting, I conluded that my NH4NITz contained a lot of sulfate, so much of the
precipitate was in fact CaSO4. I then did a lot of work trying to separate the CaSO4 from the CaNITz, but since the sulfate is not that insoluble in
water, it proved to be maybe more trouble than it was worth.
So, at this point I have aggressively recrystallized my NH4NITz to try to remove all of the sulfate. We'll see how that has worked, but if it hasn't,
it may be that it is best to extract the H2NITz from the diluted nitration mix, or possibly first precipitating with ammonia, then freeing the acid
with hydrchloric or nitric and then extracting.
|
|
|
Microtek
National Hazard
Posts: 920
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I succeded in ironing out the kinks in the nitriminotetrazolate preparation, and can now reliably produce both free nitriminotetrazole and ammonium
nitriminotetrazolate. I have also prepared CaNITz and recrystallized it. On heating at 200 C (no vacuum) exactly the calculated amount of mass,
assuming pentahydrate to anhydrous, was lost. The product seems like it should work very well, making small holes in Al foil whether heated from above
or below, but it is just no very effective at initiating PETN in a cap. I have not been able to get it to fire with 30 mg.
I found a paper titled "Development and performance evaluation of green primary explosives for use in electro-explosive devices and detonators" where
CaNITz is tested along with DBX-1 and some 4-ATRI copper complexes. CaNITz was found to be hygroscopic, and their sample regained mass corresponding
to two molecules of water at ambient conditions. This probably makes it less useful, and coupled with the rather lower than expected performance, I am
turning back to the nitrotetrazole complexes.
[Edited on 1-3-2025 by Microtek]
|
|
|
Microtek
National Hazard
Posts: 920
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I succeded in ironing out the kinks in the nitriminotetrazolate preparation, and can now reliably produce both free nitriminotetrazole and ammonium
nitriminotetrazolate. I have also prepared CaNITz and recrystallized it. On heating at 200 C (no vacuum) exactly the calculated amount of mass,
assuming pentahydrate to anhydrous, was lost. The product seems like it should work very well, making small holes in Al foil whether heated from above
or below, but it is just not very effective at initiating PETN in a cap. I have not been able to get it to fire with 30 mg.
I found a paper titled "Development and performance evaluation of green primary explosives for use in electro-explosive devices and detonators" where
CaNITz is tested along with DBX-1 and some 4-ATRI copper complexes. CaNITz was found to be hygroscopic, and their sample regained mass corresponding
to two molecules of water at ambient conditions. This probably makes it less useful, and coupled with the rather lower than expected performance, I am
turning back to the nitrotetrazole complexes.
[Edited on 1-3-2025 by Microtek]
[Edited on 1-3-2025 by Microtek]
|
|
|
| Pages:
1
..
23
24
25 |
|









