I made no attempt to buffer the reaction mixture, the large amount of acetic acid was merely to act as a solvent for the paracetamol but the pH would
still have been >3 and so within the lower limit, admittedly only just. I agree that a buffer to give a pH closer to the mid-range would have been
better but this was intended to be the first of a series of experiments. Unfortunately I have been sent overseas again and will not be home for
another 6 weeks.
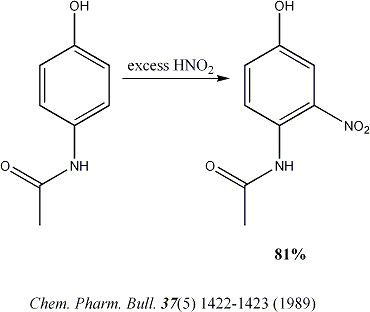
Thinking about things again the large excess on nitrous acid make no sense. My experience of phenolic nitrosation reactions is that they are very
rapid and the subsequent oxidation is slow. So with the addition of a more potent oxidizing agent the addition of more than one equivalence of sodium
nitrite is un-necessary. In fact my experience is that excess nitrous acid almost always results in the generation of much tarry material in any
reaction and my experience, as posted above, with this reaction seems to bear that out. |



 so waving a pickle over the beaker while breathing across it gently might be enough to do it.
so waving a pickle over the beaker while breathing across it gently might be enough to do it. . I think I stick with my system
given above because the addition of sodium nitrite will have its own buffering effect too.
. I think I stick with my system
given above because the addition of sodium nitrite will have its own buffering effect too.