
| Pages:
1
2
3
..
7 |
The_Davster - 15-11-2006 at 19:36
There has been talk of tetrazoles and precursors in several threads, usually as off-topic discussion, I thought it would be an idea to do a bit of a
collection of some of this into a single thread.
The 'My favorite Primary Explosive' thread on E&W, where the nitrotetrazoles are first mentioned.
http://www.roguesci.org/theforum/other-explosives/3158-my-fa...
Ironically, I found this thread after I had done all my research, which as Rosco posted all the patent refs in that thread, would have made my life
much easier had I found that thread before I dug through the literature looking for refs to the patents.
5-ATZ
US patents 5424449 and 5594146 detail the synthesis of 5-ATZ from hydrazine, cyanamide, nitrite and ammonia, but as aminoguanidine is an intermediate
in the synthesis, one can start with it. I have read that aminoguanidine hydrochloride is used in some pills that are supposed to make you live
longer, and in my opinion is much preferable to the use of hydrazine . I have
not looked further into the use of such antiaging pills as I have reagent aminoguanidine. I had worked out the ammounts of aminoguanidine needed to
pick up the procedure in the middle, but I lost my calculations, and then found what Microtek had already done it, so from there on I used his
numbers. (Much thanks, I hope you do not mind me posting your procedure)
. I have
not looked further into the use of such antiaging pills as I have reagent aminoguanidine. I had worked out the ammounts of aminoguanidine needed to
pick up the procedure in the middle, but I lost my calculations, and then found what Microtek had already done it, so from there on I used his
numbers. (Much thanks, I hope you do not mind me posting your procedure)
| Quote: |
- 6.45 g aminoguanidinium bicarbonate was suspended in 7 mL H2O and neutralized with 5 mL 30 % HCl ( dropwise addition although it cooled down rather
a lot during addition ).
- pH was checked with universal indicator strips and adjusted to ca pH 4.
- Another 5.3 mL 30 % HCl was added all at once.
- A soln of 3.5 g NaNO2 in 7.5 mL H2O was added from an addition funnel while stirring vigorously and keeping temp at 17-20 C with an ice bath ( no
salt ). Addition proceeded smoothly with negligible evolution of NOx ( not visible and only just detectable by smell ) until the very last drops which
gave the evolution of NOx which is typical of nitrite with acid. This likely signified that all the aminoguanidine had been diazotized.
- Stirring was continued for 20 minutes at 20 C after addition was completed.
- 4.9 mL 25 % ammonia soln was added all at once ( pH was measured at this point and was found to be ca 6-7 ), the flask was equipped with a condenser
and the mix was heated to beginning reflux. It was held there for 2 hours.
- While still hot, pH was adjusted to ca 4 with HCl and ammonia water; ca 1.8 mL 30 % HCl was required.[I found I actually had to basify instead at
this step]
- Mix was allowed to cool very slowly by turning off the hotplate but letting the flask stay on it. Once it had cooled to room temp, it was placed in
the refridgerator until a temp of 10 C had been reached.
The product crystallized in very well defined plates beginning from ca 40 C.
- Almost all the mother liquor was easily decanted from the dense crystal mass, and 12 mL of H2O was added to remove co-precipitated chloride.
- Mix was heated to 45 C with stirring and was then allowed to cool to 10 C.
Not all of the product was dissolved in this operation.
- Product was removed by filtration and washed several times in the filter with cool water. |
Works Great! Even did a melting point(or rather, decomposition point). The crystals are quite nice, but the solution tends to super-saturate(4/5
times), so introduction of a seed crystal at around 50C is a good idea. I added one at 60-70C once, and the resultant crystals looked different, but
still had the same melting point. The crystals formed when a seed crystal is introduced at this temp do not look as pretty, and they hold more water
taking longer to dry, so 40-50C seems ideal.
Nitrotetrazole(ate)
US patents 4093623 and 2066954 describe the prep of acid copper nitrotetrazolate from 5-ATZ, which can then used to make other nitrotetrazolate
compounds via metathesis. Example 6 of 4093623 was carried out on a scale just slightly greater than testtube scale, giving a precipitate of
CuHNT(NT)2, I did not let this dry as it is said to be sensitive to shock and electrostatic discharge, and my winters are very dry. The acid copper
salt was reacted with NaOH solution as per example 8 in the same patent, however I am simply using a slightly acidic solution of NaNT (pH=4, as per
patent directions) instead of evaporating to get sodium nitrotetrazolate crystals. However the solution has turned a light yellow/green over a couple
days, so it may be advantageous to keep it basic and then acidify before using the sodium salt to make other metal nitrotetrazolates.
Speaking of which, I prepared a very small ammount of silver nitrotetrazolate, which simply leaves me in awe. It reminds me of silver fulminate in
terms of how small an ammount of it will go DDT, even when wet. (used flame to do this). Something silver azide does not do as well. I believe
Rosco calls this an "unequivocal" primary. Sensitivity is said to be relatively low, but I have done no testing of this, and likely will not. This
was more of a long wonderful chemical adventure than a search for an ideal primary. I just decided to expend so much effort into getting to this point as I really adore the structure of tetrazoles.
Others which I have no experience with to date
Azidotetrazole:
https://sciencemadness.org/talk/viewthread.php?tid=5129
JoPEP,30, 2005, page17
Looks like crazy stuff, I should try it sometime
Nitraminotetrazole:
Can be done with either making the nitrate of ATZ, then dehydrating, or by using nitraminoguanidine in the tetrazole reaction. Unfortunatly
nitraminoguanidine requires hydrazine.
Thermochemica Acta 384 (2002) 113-120
ATZ-nitrate:
PEP, 30 (2005) No3 191
5-halotetrazoles:
Been meaning to research these, I want to make iodo and bromo
I intended to write more under this section, but I am tired now, more later perhaps. There is also a complete route from cyanuric acid(pool chlorine
stabilizer) to tetrazoles, I know Rosco is doing some work on this as well, but I intend to put a bunch more annoying steps in to avoid the use of
hydrazine. But really I have only started this part, and that would be for another thread.
I tried to attach a big review of tetrazole chemistry, but it is too big, and rapidshare is full curently. I'll post it when there is more room on
rapidshare
[Edited on 16-11-2006 by The_Davster]
DeAdFX - 15-11-2006 at 21:55
Phenyl cyanide + NaN3 + NEt3-HCl is supposed to yeild phenyl tetrazole. Since I do not know of good place to upload stuff I will send you the link
discussing the synthesis.
Tetrazylazides
Rosco Bodine - 17-2-2007 at 18:51
Liquid or plastique primaries are virtually unknown .
Ethyleneditetrazylazide is reportedly a way to do this .
I do not know of any information available concerning
synthesis of these tetrazylazides .
Attachment: US2170943 Tetrazylazides.pdf (80kB)
This file has been downloaded 3077 times
The_Davster - 18-2-2007 at 00:19
Is that archaic nomenclature? Could tetrazyl be the old name for tetrazoyl? Making the compound in question an 'ionic liquid' of whatever 2
molecules of 5-ATZ linked through nitrogens to the NH2s on ethylene diamine is called, then protonated somewhere and the azide salt formed?
Or it could be a di substituted tetrazole ring...but I can't imagine a bridging ethylenediamine stabilizing any azotetrazole.
Really, I am very intrigued!
Rosco Bodine - 18-2-2007 at 00:50
I don't understand the term tetrazyl either ....
wonder if it is equivalent to " tetrazolyl " ?
The germans used a different nomenclature
and it is a bit confusing . The same thing occurs
with regards to some of the other tetrazole patents .
IIRC there are a few lines in COPAE about some
of the different synonyms used for the same groups .
Defintely encounterd this nomenclature confusion before ,
and it seems that it was Axt or Nick F who was good at sorting these out .
Anyway , those are reputable names on the patent ,
company and inventor alike ....so there is likely something
to this group of compounds , which they aren't saying
too much about with regards to synthesis .
The attached file sheds some light on this ......
whew what a synthesis , reserved for the skilled
and not squeamish about toxic materials ....
[Edited on 18-2-2007 by Rosco Bodine]
Attachment: US2179783 Tetrazylazide and Salts.pdf (103kB)
This file has been downloaded 2749 times
Rosco Bodine - 18-2-2007 at 01:13
Here is more information expanding on that first patent
Attachment: GB510992 Tetrazylazide Priming Explosives.pdf (155kB)
This file has been downloaded 3145 times
The_Davster - 18-2-2007 at 12:07
From what I gather...The first German patent is only claiming an improved process for mass production of the azotetrazole...For which there are other
known routes too, not involving cahones as big as would be necessary for that patent
So now we are definatly getting somewhere...it is an azotetrazole. Excellent. Those are not impossible. However the ethylene part of the molecule
is still confusing.
Unfortunatly, due to the standard synths of disubstituted tetrazoles, azotetrazole probally cant be used as the starting material. Unless its an
ethyl-azo disubstituted tetrazole, in which silver azotetrazole and ethyl iodide should give the product.
I wish the patent had a molecular structure for the compound...
Rosco Bodine - 18-2-2007 at 12:47
Looks to me like all that is required for the ethylene derivative is to react ethylene dichloride with 2 equivalents of sodium tetrazylazide .
The basis for these salts is Tetrazylazoic Acid ....
which seems to be hydrazoic acid which has
been combined with a tetrazole ring ......
now that should enhance the already incredible
energy and also lend a greater stability if my guess
is correct .
I have to wonder if the sodium tetrazylazide could be substituted for sodium azide in the azo-clathrate synthesis leading to a series of
tetrazylazo-clathrates which would tame the overly sensitive straight lead tetrazylazide ...while manifesting extraordinary power .
My guess would be that the critical diameter on these tetrazylazides would be extremely small ....capillary sized
det cord anybody ? Did somebody ask for explosive paint ?
This stuff should do the trick
[Edited on 18-2-2007 by Rosco Bodine]
The_Davster - 18-2-2007 at 13:11
Oops, yeah, meant diiodoethane last post.
I don't know, I am a bit unsure whether what bridges the two tetrazoles is a -CH=CH- or a simple -CH2-CH2-. Archaic nomenclature can be a bit strange
for such things. Either way, makes me very curious about what would happen with the -C(triplebond)C- variant, but dichloroacetylene as a starting
point is... . Diiiodo would also probably work...
. Diiiodo would also probably work...
Hmm, I have some sodium nitrotetrazole crystals lying around, and diiodoacetylene is easyish to make(but rather toxic for my tastes)...I might just
have a go on a variant like this...
Should be safer than azotetrazole variants, as I have heard horror stories about azotetrazole. NaNT at least turns to a liquid before it dets with
surprising violence.
(Rosco, thanks for bringing these up from obscurity, I had been in a chemistry slump and nothing was really catching my attention, these brought me
out)
As for a substitute for NaN3 in the azo-clathrates...Why not use both azotetrazole, and azide. With some nitrotetrazole added to taste
[Edited on 18-2-2007 by The_Davster]
Rosco Bodine - 18-2-2007 at 13:24
A cauldron full of complexes
is kind of like
a saucer full of secrets .....
What you get
might even trump
old Forrest Gump
with life that's like
a box of chocolates
You never really know
exactly what you are going to get
I would add that a healthy dose of paranoia
would not be out of place concerning this class
of energetic materials ....I'd definitely keep quantities
very small , recognizing that this sort of material is
right up there among the most energetic materials
known to science . It wouldn't take much of this stuff
doing the unexpected to do a lot of damage .
[Edited on 18-2-2007 by Rosco Bodine]
Rosco Bodine - 18-2-2007 at 22:57
I was right in thinking this " tetrazyl " prefix is perhaps equivalent to " tetrazolyl " , and have been able to further clarify exactly what it is ,
as well as a possibly easier method of synthesis .
The same compound having the following *six* synonymous designations is described in PATR Vol.9 , T-124, 125
5 - Azido Tetrazole , Tetrazolyl Azide , 5 - Triazo - Tetrazole ,
Diazotetrazolimide , Tetrazylazoimide ,
[ Tetrazolyl- (5) ] - azoimide
It is also item #2 on page 2 of the attached patent .
In COPAE , page 447 , Davis describes in more detail the method mentioned in the patent , using tetracene as the precursor , hydrolyzed using NaOH ,
to form an intermediate triazonitroso-aminoguanidine which is isolated by addition
of copper acetate producing a precipitate of bright blue colored explosive copper salt . This salt after filtering and washing and treatment with
acid ( probably HCl ) gives
the Tetrazolyl Azide .
So essentially , Calcium Cyanamide , Hydrazine Sulfate ,
and sodium nitrite ......or Aminoguanidine Bicarbonate and
sodium nitrite are the precursors for tetracene , and this
eliminates the need for more extreme and toxic cyanogen and azide reagents proposed by the Friedrich patent .
Davis cites a reference providing more on this and I will be requesting it through channels here .
The Chemistry of Aminoguanidine and Related Substances ,
G.B.L. Smith , Chem. Rev. , 25 , 214 (1939)
Attachment: GB185555 Tetrazole Initiator Compounds.pdf (286kB)
This file has been downloaded 3077 times
The_Davster - 19-2-2007 at 01:16
There is also a route from 5-ATZ to azidotetrazole, I have it hardcopy and will dig it up hopefully. Just need to find my folder on the
tetrazoles...its around here somewhere.
Looks fun: https://sciencemadness.org/talk/viewthread.php?tid=5129
I also have JoPEP,30, 2005, page17 written down as a ref for them.
But for now, uploaded is the chemistry of aminoguanidine file you wanted:
I should move this out of the complex thread and into the tetrazoles thread I started a while back. Fine with you? These arent really complexes....
EDIT: Crap, too big to attach here, I rapidshare.COM ed it
http://rapidshare.com/files/17182917/The_Chemistry_of_Aminog...
[Edited on 19-2-2007 by The_Davster]
The Chemistry of Aminoguanidine and Related Substances
Rosco Bodine - 19-2-2007 at 02:05
Here's a smaller file .....
Thanks to kmno4
And yes any alternate routes are of interest .
Actually there are only a few of these
tetrazoles of particular interest as most of them
have been evaluated and ruled out for
oversensitivity or other reasons .
Funny you should post that link for the thread
about the acetic acid ....and yeah I was thinking about
what was mentioned about the incompatability ,
realizing that the copper precipitate of the nitroso
derivative of tetracene must be formed in alkaline solution ,
and then separated from the sodium acetate in alkaline
supernatant and filtrate , carefully rinsing out any acetate
from the copper precipitate , *before* acidifying .....
or else a nasty surprise awaits
It would really be nice to have a more forgiving method ,
which doesn't involve copper salts ....maybe a calcium salt
for example .
[Edited on 19-2-2007 by Rosco Bodine]
Attachment: cr60081a003.pdf (975kB)
This file has been downloaded 5326 times
5-azidotetrazole via Ba(OH)2 hydrolysis of tetracene
Rosco Bodine - 3-3-2007 at 20:04
Thanks to Joeychemist and solo for this helpful reference .
See the last paragraph on page 5 ,
Alkaline degradation of IX (tetracene monohydrate) .
Evidently simply boiling tetracene with a base hydrolyzes
it with the expulsion of one ammonia , and leads to
ring closure and formation of 5-azidotetrazole .....perhaps
(hopefully) being present as the soluble 5-azidotetrazole salt of sodium , in the case where the base used for the hydrolysis is sodium hydroxide .
This should simplify greatly
the synthesis of 5-azidotetrazole where it is needed as
an intermediate which will not be isolated , knowing
that it is produced *directly* by the alkaline hydrolysis of
tetracene .
This finding is in contrast with what Davis reported on COPAE concerning the incorrectly alleged intermediate triazonitroso-aminoguanidine identified
mistakenly by Hofmann via elementary analysis , whose composition was actually identical with the monohydrate copper salt of
5-azidotetrazole .
An interesting side note concerning the related derivative
of tetracene , 5-aminotetrazole . I have read in PATR ,
tetracene quietly decomposes / converts completely to
5-aminotetrazole by nothing but gentle heat being applied to the sample of tetracene for several days ,
@90C less than 3 days .
[Edited on 4-3-2007 by Rosco Bodine]
Attachment: The Structure of Tetracene.pdf (778kB)
This file has been downloaded 3469 times
The_Davster - 4-3-2007 at 00:04
Excellent!
For the aminotetrazole though....90C for 3 days? Reflux diazotized aminoguanidine in ammonia for 3h...82% yields. Tried and true.
Rosco Bodine - 4-3-2007 at 01:11
Yeah it is in PATR under " Tetrazene " , an Australian
government study found that stab detonators containing
tetracene would fail to function after storage at elevated temperatures because of the tetracene slowly changing to
5-aminotetrazole by thermal decomposition . They stated
complete conversion at 90C in less than 3 days .
With regards to the reaction of the 5-azidotetrazole sodium salt with methyl or ethyl sulfate or chlorides ,
I wonder if other esters such as the nitrate or nitrite
esters would work as well , or if the nitrite ester might
lead to something different . I especially wondered about the reaction possibility for the nitrite esters since they are so easily made .
Rosco Bodine - 8-3-2007 at 18:47
There is probably a workable general route to the useful energetic tetrazoles and related materials
which uses OTC precursors .
The production of Calcium Cyanamide or Zinc Cyanamide
or Magnesium Cyanamide ....or perhaps some mixture
of these cyanamides which might form more readily by
pyrolytic decomposition of their cyanurate precursors ,
could then be reacted with Hydrazine Sulfate to form
aminoguanidine as the intermediate ....which is then
reacted with sodium nitrite or perhaps nitrosated ,
" diazotized " by other methods , and depending upon the
pH at which this is done , the product is guanyl azide ,
or tetracene , or 5-aminotetrazole ......by variations upon
the process .
Guanyl Azide Picrate is itself another useful , stable and powerful energetic material
which is satisfactory for use as a base charge in detonators .
To any of these products then can be applied subsequent treatments
to obtain the desired tetrazole compound .
IIRC , the Zinc Cyanurate is the material which is converted at a somewhat lower temperature
to the desired cyanamide . It still requires a low red heat ,
but this should be reachable without too much difficulty ,
in a rudimentary sort of furnace , or even in an open
fire or over a burner .
A preliminary experiment I did seemed to produce the desired cyanurate precursor ,
but I haven't yet fired this material to see if the cyanamide is produced as expected ,
however it should work as this reaction is mentioned in several patents .
https://sciencemadness.org/talk/viewthread.php?tid=2762&...
Tetracene can be produced from the reaction of the
alkaline earth cyanamide and hydrazine sulfate ,
without isolation of the aminoguanidine intermediate ,
by treatment of the reaction mixture with NaNO2 .
I have a patent concerning this and I will find it
and attach it later .
I expect there are perhaps a dozen different energetic materials which are of reported value ,
maybe half of those of special interest among this class of compounds .
[Edited on 8-3-2007 by Rosco Bodine]
The_Davster - 8-3-2007 at 19:26
I actually made a dozen grams or so of the calcium cyanurate. In another post you say it decomps to HOCN. That should instantly combust at the
required temps hopefully right? No toxic death cloud?
[Edited on 8-3-2007 by The_Davster]
Rosco Bodine - 8-3-2007 at 20:18
Right ...cyanic acid should burn off , and then extinguish as
the excess cyanic acid comes off at a lower temperature stage of the decomposition where the cyanurate
is probably converted first to a straight cyanate .
The evolving cyanic acid vapor might not ignite within an
electric furnace being gradually heated ....but it should ignite in a flame fired environment for sure . If you are using a
metallic vent line , simply discharging it onto the flame of a small burner used as a pilot light should flare it off okay .
If you wanted to bubble any offgassing into alkaline water
you should use a trap or checkvalve to prevent any suckback of the liquid when the rate of gas evolution slows .
IIRC , The cyanuric acid is actually a trimer of cyanic acid ,
thermally decomposed .....a parallel sort of material
as is trioxane and formaldehyde . Anyway , after the
alkaline earth cyanurate is decomposed to its normal
cyanate ....the "extra" HOCN is gone ....and further
decomposition as the material reaches red heat ,
evolves only CO2 ....and when it finishes offgassing
CO2 .....all that should be left is pure white Calcium Cyanamide .
Of course this would not be a commercially viable process because it is counterintuitive to sort of reverse engineer
the usual products of a process along a tangent to get back to an intermediate
....
in our case the alkaline earth cyanamide .
But for a lab scale approach to how easiest to take an
OTC material like chlorine stabilizer , cyanuric acid , and
produce pure calcium cyanamide or other pure cyanamides from it ....this would seem the most probable
candidate as a method , doing a straightforward thermal
decomposition of an intermediate which should produce
nothing else but the desired product .
Attached is a patent describing the conditions for reaction
of calcium cyanamide with hydrazine sulfate to form aminoguandine in solution , and a filterable form of
calcium sulfate residue by control of pH which influences the
crystalline form of the of the calcium sulfate byproduct .
The aminoguanidine solution in this patent process is
then neutralized with H2SO4 to form a solution of aminoguanidine sulfate , to which is added baking soda
to precipitate insoluble aminoguanidine bicarbonate which
is filtered out .
But you see at the point where the aminoguanidine freebase
or sulfate is available in aqueous solution , other things can be done to obtain different products if aminoguanidine bicarbonate is not the desired
end product .
Guanyl Azide or its salts , or tetracene , or 5-aminotetrazole ,
or other materials may be obtained by further reactions upon the aminoguanidine intermediate without any need for its isolation .
[Edited on 9-3-2007 by Rosco Bodine]
Attachment: US3673253 Aminoguanidine Bicarbonate via Calcium Cyanamide and Hydrazine Sulfate.pdf (385kB)
This file has been downloaded 3348 times
5-Nitroaminotetrazole from 5-Aminotetrazole
Rosco Bodine - 9-3-2007 at 21:18
The 5-aminotetrazole nitrate dehydration to form
the 5-nitroaminotetrazole has been described as
a somewhat analogous reaction to the dehydration of
guanidine nitrate to form nitroguanidine . The conditions must be carefully controlled and the temperature kept low due to instability of the amino
group of the amino tetrazole , which will be oxidized under too severe conditions . The free acidic 5-Nitroaminotetrazole should
probably not be isolated as the dry material but left in solution , or converted to one of its more stable salts .
There are a few ways of doing the conversion directly
from 5-Aminotetrazole , not requiring its nitrate as
an intermediate . Attached is a patent which describes
5-Aminotetrazole being dissolved in concentrated H2SO4
and then nitrated with fuming (90%) HNO3 added dropwise @20-25C , the mixture drowned over crushed ice and then neutralized with NH4OH to precipitate
the
diammonium ? salt of 5-Nitroaminotetrazole . Other salts
can be made from this . See page 4 , Column 5 ,
Example 1 .
Alternately to using the mixed acid , a Japanese patent
JP11279164 describes that the nitration can be accomplished by addition of NH4NO3 to the solution of
5-Aminotetrazole in concentrated ? H2SO4 . Since I
am unable to read Japanese ....the details of the method
are unclear . But it appears that the process is otherwise
similar .
According to the reactions shown in the Japanese patent
and also other English language references , the free acidic 5-Nitroaminotetrazole is obtained in solution by treatment of the ammonium or sodium salt
with HCl .
The potassium salt is the salt specified for use in the reaction for producing " Stabanate " , the double
lead salt of 5-Nitroaminotetrazole and Styphnic Acid ,
which is a patented US3310569 initiator having superior stability and 50% higher brisance in the sand test than lead styphnate . It is reportedly
satisfactory for use as
an initiator in detonators , although its performance in that regard is not disclosed .
[Edited on 15-3-2007 by Rosco Bodine]
Attachment: US5516377 5-Nitroaminotetrazole by nitration of 5-Aminotetrazole.pdf (521kB)
This file has been downloaded 2955 times
Here's an older related journal reference
Rosco Bodine - 9-3-2007 at 22:01
The Nitration of 5-Aminotetrazole
Attachment: The nitration of 5-aminotetrazole.pdf (398kB)
This file has been downloaded 2998 times
Sobrero - 10-3-2007 at 08:43
I have prepared 7.25g nitroaminoguanidine as follows: (Patent US2617826)
(hydrazine sulfate was made with Mr. Anonymous's recipe, nitroguanidine was made from homebrew guanidine nitrate)
200ml water was heated to 60�C and 11g NQ were added while stirring. 15.3g hydrazine sulfate, 100ml water and 6.90g NaOH were mixed and stirred
until everything had gone into solution. This solution was slowly poured into the agitated and heated NQ-solution/suspension. Heating (60�C) and
stirring was continued during half an hour. After 15 minutes all the NQ had dissolved. The liquid turned orange and during the experiment I smelled
ammonia:
HN=C(NH2)(NHNO2) + H2N-NH2 ---> HN=C(NHNO2)(NHNH2) + NH3
So after these thirty minutes, the solution was neutralised with concentrated hydrochloric acid (approx. 6ml HCl 29% was used). The liquid was allowed
to cool to 5�C (fridge) and was left so for one hour for full precipitation of the NAQ (the colour... how should I say: slightly pale yellow -
"blanc cass�" - light off-white colour?), . It was then filtered and washed with two portions of 15ml cold distilled water. Dried at 80�C.
Yield: 7.25g or 57.6% yield.

BTW: I added two teaspoons of sodium nitrite and 10ml GAA to the filtrate. A very light foaming occurred (no smell or colour, I gess it's nitrogen)
and after a few hours I noticed a white somewhat fluffy precipitate. I filtered it, washed it with some water and let it dry. Unfortunately it only
burns (no deflagration or something energetic). Does anyone have an idea what this stuff is?
Now, can someone give some help for the synthesis of nitroaminotetrazole and nitroguanyl azide (starting from NAQ off course). The only documents I
found was Reaction of nitrous acid with nitroaminoguanidine and Ultraviolet absorption of 5-NATZ and it salts but as can be seen no specific reaction conditions, weights, procedure, ... are given. PATR (page
A259-260) is not very helpfull too  .
.
[Edite le 10-3-2007 par Sobrero]
Rosco Bodine - 10-3-2007 at 13:08
(Edit)
Nitroaminoguanidine is evidently an alternate precursor for nitroaminotetrazole .
The other path would be to reduce the nitroguanidine using zinc , to get aminoguanidine , and then nitrosation , followed by cyclization to
5-aminotetrazole and nitration as described earlier .
The entire article may provide some additional information
concerning the alternate route described in the abstract
which you just linked for nitroaminotetrazole from
nitroguanylazide . I will request that article .
( Edit : See complete article attached in following post )
Nitroaminoguanidine can be nitrosated to form nitroguanylazide .....by a similar method as is used
for conversion of aminoguanidine to guanyl azide .
The following excerpt US6350307 , page 9 , column 15 ,
line 30 , concerns guanyl azide
Preparation of Guanyl Azide
An initial charge of technical grade hydrochloric acid and distilled and demineralized water is indirectly cooled down to about 0.degree. C.
Aminoguanidine bicarbonate is subsequently added at about 10.degree. C. in the course of about 30 minutes. Thereafter, the batch is indirectly cooled
down to about 0.degree. C., and sodium nitrite solution is added up to max. 15.degree. C. in the course of about 3 hours. This is followed by stirring
with nitrite excess for 15 minutes. Directly prior to transfer of the azo groups, the nitrite excess is removed with amidosulphonic acid.
A Ukranian patent (attached) describes this , including isolation of the product . There is an English summary on page 2 .
Abstract of UA16960U
A method for producing nitroguanilazide and derivatives thereof comprises nitrosation of N-nitro-N'-aminoguanidine (NAG) in the aqueous medium in the
presence of sulphuric acid by potassium nitrite at a molar ratio of and providing water excess of 1.48 l H2O/mole of NAG at the temperature of with
subsequent keeping the reaction mixture at the room temperature during specified time, evaporation of reaction mass during a long time at the same
temperature, three-stage extraction of solid remainder by anhydrous diethyl ether at the amount of 1.2 l (C2H5)2O per 1 mole of output NAG,
evaporation of ether remainder and obtaining nitroguanilazide (NGA) with an output of 77 %. At that for nitrosation nitrites of alkaline
andalkaline-earth metals - MNO2 (M=Na, Li, K, 1/2Ca, 1/2Mg, 1/2Ba, 1/2Sr) are used at a molar ratio of as acid H2SO4, HNO3, H3PO4, HBr are also used,
it is carried out at cooling, the reaction mass is filtered and cooled, keeping it in these conditions for 5-10 hours, the residue is filtered, dried
and nitroguanylazide is obtained with an output of 40-55 %, and aqueous solution after filtration of NGA is extracted by diethylether at an amount of
the extract is evaporated and additional amount of NGA is obtained (15-40%).
But I would not recommend isolation of
the nitroguanyl azide , as much as I would personally
have interest in the possible picrate , given that the
analogous (non-nitro) guanyl azide picrate is already known
and has been observed to be stable , and useful as a base charge for detonators . The possibility exists that the
Nitroguanyl azide picrate , having its additional nitro
group would also be interesting , and hopefully have adequate stability along with greater power than the
already known " ordinary " guanyl azide picrate .
[Edited on 15-3-2007 by Rosco Bodine]
Attachment: UA16960 Nitroguanylazide Ukranian patent.pdf (279kB)
This file has been downloaded 2541 times
Nitrosation products of Nitroaminoguanidne
Rosco Bodine - 10-3-2007 at 20:07
The Reaction of Nitrous Acid with Nitroaminoguanidine
Here is the entire article associated with that
single page linked above .
Attachment: The Reaction of Nitrous Acid with Nitroaminoguanidine.pdf (435kB)
This file has been downloaded 3378 times
Rosco Bodine - 13-3-2007 at 22:21
Here's an aminotetrazole related primary
Attachment: US3663553 Di-Silver Aminotetrazole Perchlorate.pdf (101kB)
This file has been downloaded 2970 times
more metal complex salts
Rosco Bodine - 14-3-2007 at 14:26
This patent has a few interesting complex salts ,
and also provides some citations of references
which may be of more general interest .
Attachment: US5962808 Metal Complex Oxidizer Salt Gas Generants.pdf (538kB)
This file has been downloaded 3251 times
5,5'-Diazoaminotetrazole
Rosco Bodine - 14-3-2007 at 18:14
Also called
Bis(tetrazolyl)-5)-triazene ,
1,3-Bis(1H-tetrazolyl-5)-triazene ,
I,III,-Di-(tetrazolyl-5)-triazene ,
1,3-Di-[tetrazolyl-(5)]-triazene
The sodium salt is obtained by nitrosation of aminoguanidine
dinitrate or aminoguanidine sulfate , by dropwise treatment with NaNO2 solution added to a sodium acetate and acetic acid buffered solution of the
aminoguanidine salt at 15C .
5-aminotetrazole by the same treatment gives the same product . The sodium salt is reportedly not explosive ,
but leads to other metallic salts by metathesis , which are explosive . The lead salt , and the lead double salt with
lead styphnate are particularly interesting .
See PATR vol. 9 page T-121
Lead-5-5'-Diazoaminotetrazolate Styphnate is reportedly
an " ideal replacement " for lead azide .
[Edited on 19-3-2007 by Rosco Bodine]
Attachment: US2064817 Manufacture of Diazotized Tetrazole Derivatives.pdf (98kB)
This file has been downloaded 2619 times
Rosco Bodine - 14-3-2007 at 18:18
US2090745 describes the single
Lead 5-5'-Diazoaminotetrazole salt
[Edited on 14-3-2007 by Rosco Bodine]
Attachment: US2090745 Lead 5-5'-Diazoaminotetrazole.pdf (93kB)
This file has been downloaded 2621 times
The " Stabanate " patents
Rosco Bodine - 14-3-2007 at 18:30
One of the useful things which can can be done with
nitroaminotetrazole
US3310569
Crystalline Double Salt Pb Styphnate and Pb Nitroaminotetrazole
GB1069440 British patent same subject
Attachment: US3310569 Stabanate.pdf (506kB)
This file has been downloaded 2460 times
Azotetrazole , 5,5'Azotetrazole
Rosco Bodine - 15-3-2007 at 00:05
See PATR , Vol.1 , A-569
The sodium salt is produced in 76% yield from a solution of
5-aminotetrazole in 15% NaOH at 50C by oxidation with KMnO4 solution added dropwise . Unreacted KMnO4 is
decomposed with alcohol , and the solution is refluxed for 1 hour at 100C . On cooling the sodium salt is obtained as
crystals .
Attachment: US5877300 5-5'azotetrazole and derivatives.pdf (153kB)
This file has been downloaded 2792 times
Lead Azotetrazole
Rosco Bodine - 15-3-2007 at 00:29
Here is a British munitions patent for the
lead salt of 5,5'-Azotetrazole
Attachment: GB986631 Lead Azotetrazole.pdf (176kB)
This file has been downloaded 2618 times
5-Nitrotetrazole
Rosco Bodine - 16-3-2007 at 13:42
This is one of the energetic tetrazoles which is a bit trickier
to make because of an unstable intermediate diazo compound involved in its formation .
A special technique has been devised for avoiding explosion of the unstable intermediate which otherwise is problematic , and this special method has
been further refined and described in a subsequent patent .
[Edited on 16-3-2007 by Rosco Bodine]
Attachment: US2066954 C-Nitrotetrazole Initiator Compounds.pdf (338kB)
This file has been downloaded 2936 times
5-Nitrotetrazole improved method
Rosco Bodine - 16-3-2007 at 13:47
This patent studies and refines the synthesis of
5-Nitrotetrazole , improving the safety and yields .
Attachment: US4093623 Copper Salt intermediate for 5-Nitrotetrazole.pdf (469kB)
This file has been downloaded 2827 times
Copper Ammonium Salt of Diazoaminotetrazole
Rosco Bodine - 16-3-2007 at 14:44
While on the subject of copper salts of tetrazoles .....
Here's an interesting one which I haven't run across before
It seems possible that similar ammonium salt could be possible for silver , zinc , iron , cobalt , chromium and nickel .
[Edited on 16-3-2007 by Rosco Bodine]
Attachment: US2004719 Copper Ammonium Salt of Diazoaminotetrazole.pdf (190kB)
This file has been downloaded 2705 times
The_Davster - 16-3-2007 at 15:18
Ok threads merged.
Rosco, thanks for posting all the patents and articles, I know I made mention of a few in the first post, but having them all in one spot online is
really conveinent.
" Green Primaries "
Rosco Bodine - 16-3-2007 at 15:33
There is a double salt formed from either sodium or ammonium nitrotetrazole , combined with either copper or iron nitrotetrazole as a stable dihydrate
.
These compounds are presently being investigated as
relatively non-toxic replacements for lead azide and lead styphnate containing compositions which have been the
established standard primaries in use for many decades .
Attachment: Green Primaries 1.pdf (662kB)
This file has been downloaded 3114 times
Present State of the Art
Rosco Bodine - 16-3-2007 at 15:42
More of the same from Los Alamos
concerning the so called
" Green Primaries "
http://www.sciencemadness.org/scipics/Green Primaries 2.pdf (2.96 MiB)
[Edited on 25-3-2007 by Rosco Bodine]
Tetrazole Double Salts
Rosco Bodine - 16-3-2007 at 16:12
| Quote: |
Originally posted by The_Davster
Ok threads merged.
Rosco, thanks for posting all the patents and articles, I know I made mention of a few in the first post, but having them all in one spot online is
really conveinent. |
Thanks for merging these threads .
You are welcome concerning the file collection . I was making a dedicated folder of tetrazole related files and decided to share the folder with
summary notes and commentary to save others covering the same ground .
It's long overdue to collect most of the pertinent tetrazole related references in one place . I tried not to hop around too much , but to keep the
related files together in some intelligent sequence , or as close as I could keep 'em somewhat topically collated .
I still think tetrazoles deserves a sticky thread status ,
to keep from digging it up when it gets pages behind , and avoid a lot of new lookups or newcomers or the lost
posting new topics that are already right there on top in an existing thread .
They are sort of the " top dawg " energetic materials so
as such ....on top is not out of place as a place for the
topic for them to be
Anyway , back on topic ...
As a general rule it has been the case that many of the energetic tetrazoles are not satisfactory alone as initiators ,
for being too sensitive , or not having the desired crystalline form and density ....however the same tetrazole salts also
as a general rule form double salts , either combining with
other tetrazole salts as in the above described " green primaries " , or in forming basic salts , or some combined basic salts where a basic metal
tetrazole is coprecipitated
with a basic picrate or a basic styphnate . Some of these combined salts may have desirable properties which is
more than just the simple sum of the precursors properties
observed separately . There are only a few of these
combined salts reported , which I have been able to find anyway , so there could be many different possible mixed salts , or even possibly
"tetrazo-clathrates" , which have
not been reported , and some of these materials could have excellent properties as initiators . The idea is not new ,
and the attached patent gives some insight into one method of coprecipitation , where the combined solution of the sodium salts of the acidic
energetic tetrazoles and/or other acidic energetic materials is run into a solution of the lead salt or other metallic salt , resulting in a
coprecipitation of
the mixed salt . There are other strategies for the formation of mixed salts , but this is a good one as a starting point for
such experiments .
[Edited on 16-3-2007 by Rosco Bodine]
Attachment: US1580572 Tetrazole Double Salts.pdf (143kB)
This file has been downloaded 3228 times
The_Davster - 16-3-2007 at 18:35
US patent 4093623 in regards to the synth of nitrotetrazoles is the one which I found to work well, albeit a large volume and a volumous sludge of a
precipitate of the Cu-nitrotetrazole. Gravity filtration should not even be considered, and a small ammount of final yield comes from a large volume
of sludge necessitating a disporportionately large buchner funnel for filtration. This is what severly limits all nitrotetrazole synthesis that I
have read.
It appears the green types or primarys are starting to come on the market, in the form of nontoxic primers.
http://www.sellier-bellot.cz/nontox-cartridges.php?product=1...
No idea the composition though...
Rosco Bodine - 16-3-2007 at 22:01
I don't know if this has already been tried or not .
Something from the " Green Primaries 1 " development
may be applicable here , so that instead of the acid
copper salt of nitrotetrazole being the intermediate ,
which has filtration problems ....possibly the crystalline
" green primary " double salt , Na2[Cu(NT)4]-2H2O could be produced directly . If workable this would avoid having to digest the acid copper
nitrotetrazole with
caustic soda to produce the sodium salt and then subsequently reacting that with copper nitrate to form
the double salt " green primary " .
Possibly using Copper Nitrate , easily made from copper carbonate and HNO3 if necessary , and using HNO3 for the mineral acid would be better than
using the copper sulfate and sulfuric acid . These would simply be substituted in the method of US4093623 , in proportions
which would favor the formation of the double salt
" green primary " .
Running the two solutions of equal volume simultaneously and slowly in separate equal streams into the mixing and reaction vessel might give a denser
precipitate .
The completed addition mixture is held at reflux for five hours and cooled for crystallization in the synthesis reported for the green primary
Na2[Cu(NT)4]-2H2O ,
and that extended heating is possibly for crystal growth ,
as I would expect the actual chemical reaction is not
sluggish , but rapid .
I have not performed this reaction so I have no direct knowledge of this or what may happen . It very well could be that simply holding the acid
copper nitrotetrazole
precipitate of the US4093623 patent at a high temperature of say 95C or so for several hours might
cause sufficient crystal growth to produce a readily filterable product . It is understandable how there may be some reluctance to subject these
materials to digestions at elevated temperatures , for fear of
that proverb involving curiosity and cats being realized ,
as much as any expectation of improvement on a process .
However , von Herz did report conversion of the acid copper salt to the sodium salt and copper oxide , under
the condition of elevated temperature , provided by
" boiling aqueous suspension with soda lye ( NaOH) " .
See US2066954 , page 2 , line 3 .
Of course the acid copper salt had been filtered and rinsed free of excess acid prior to this treatment , but
even so this would indicate that possibly the pH could be adjusted for a
" one pot synthesis " of the copper based
" green primary " .....and also that the temperature
could possibly be raised somewhat in hopes of obtaining a more easily filterable acid copper salt by the method reported in the patent .
Another thought I had is that possibly ammonium nitrate , could be used for decomposition of the excess NaNO2
for either reaction strategy . This could possibly not work however because of the possibility of complexation of
the ammonia with the copper .
Quite a few things to ponder about this one , and only some experiments would tell the tale on these things .
Update: attached is the " patent pending " application from
the same inventors concerning several of the green primaries
[Edited on 19-3-2007 by Rosco Bodine]
Attachment: US2006030715 Complex Nitrotetrazole Primary Explosives.pdf (316kB)
This file has been downloaded 2860 times
A possible simplified method for aminoguanidine
Rosco Bodine - 23-3-2007 at 10:27
While studying these tetrazole patents ,
I found a related matter mentioned in a patent
US5041661 (attached) ,
concerning the production of aminoguanidines ,
where the use of guanidine nitrate is described
as the precursor reacted with 1, 2, or 3 moles
of hydrazine to form mono , di , or triaminoguanidine nitrate .
The reaction follows the same course essentially as when
nitroguanidine is reacted with hydrazine to form nitroaminoguanidine ......but using guanidine nitrate
leads to a non-nitro , plain aminoguanidine product .
It seems likely that aminoguanidine bicarbonate could be made and isloated from the reaction mixture of *any* soluble guanidine salt with hydrazine
sulfate having been digested for a time at the appropriate pH , then treated with CO2 or sodium bicarbonate .
If possible , this would eliminate having to convert guanidine nitrate to nitroguanidine and reduce to obtain the aminoguanidine . Reacting the
guanidine nitrate or any other soluble guanidine salt with hydrazine should give the aminoguanidine directly
The patent only describes the reaction as being applicable
for the nitrate salt of guanidine , but it would seem to me
likely that any guanidine salt should work as well .
Evolution of ammonia would be a sure sign the reaction is proceeding , if it goes as expected .
If workable this would provide a convenient alternative precursor for those who have a guanidine salt available , but do not have cyanamide .
Attachment: US5041661 Aminoguanidine , Diaminoguanidine or Triaminoguanidine via Guandine Nitrate and Hydrazine.pdf (285kB)
This file has been downloaded 2855 times
More of interest concerning triaminoguanidine
Rosco Bodine - 23-3-2007 at 11:34
It is known that aminoguanidine forms from the reaction of calcium cyanamide and hydrazine sulfate .
The inference I get from reading descriptions of these reactions , is that the mono-aminoguandine being formed
will subsequently add further amino groups to form diaminoguanidine and triaminoguanidine , in the same reaction system , depending upon the
availability of
additional hydrazine for formation of the higher ,
poly-aminoguanidines .
Dicyandiamide will reportedly depolymerize in such a reaction system , to provide the mono-cyanamide component , reacting with three hydrazines to
form triaminoguanidine .
See US3285958 attached . Possibly also the level of amination of the aminoguanidine produced is also dependant
upon the quantity of hydrazine available for reaction by
this method , and it may be possible to secure the mono-aminoguanidine nitrate , ( or other acid salt ) simply by
adjusting the quantity of hydrazine available in the reaction system to be only sufficient for the desired product .
While the monoaminoguanidine salts are of special interest as tetrazole precursors , the Triaminoguanidine salts are
stable , powerful , useful high explosives also , the nitrate and the picrate are initiator sensitive secondary high explosives , and the perchlorate
is a fuse sensitive HE primary which is probably useful as either an initiator or
as a single component charge in detonators . A chlorate
salt and other energetic salts have also been mentioned ,
but their properties have not been described in any of the references I have seen .
Attachment: US3285958 Triaminoguanidine Nitrate from Dicyandiamide and Hydrazine.pdf (298kB)
This file has been downloaded 2964 times
chemoleo - 23-3-2007 at 18:51
I just love these nitrogen-rich compound reactions!
The patent mentions the condensation of phenylhydrazine and dicyandiamide. Interestingly, dinitrophenylhydrazine is a common reagent for ketone
detection, and thus should be possible to obtain relatively easily. A condensation with dicyandiamide should give
(dinitrophenyl-NHNH)2C=N-NH-(dinitrophenyl), or possibly a nitrate salt thereof. Thus a potentially very interesting combo-energetic compound, being a
nitrate salt, containing N-N bonds, and nitrophenyl moieties! Not many EM's could claim that!
Diazoguanidine ( guanylazide )
Rosco Bodine - 25-3-2007 at 00:10
Guanylazide is also called Diazoguanidine or Azidoformamidine .
As an intermediate for 5-aminotetrazole the guanyl azide
is not isolated but is cyclized with ammonia .
Freeform Guanylazide is unstable and is only isolated as its stable salts .
The guanylazide picrate has been mentioned earlier
on the first page of this thread . It has been patented US2125462 ( attached ) for use as a base charge in detonators .
Possibly the styphnate could also be interesting ,
as styphnic acid could bond with *two* of the guanylazide
groups and unless it tended towards formation of a hydrate ,
it should be more powerful than the picrate . It would also seem possible the formation of a basic lead guanylazide styphnate , and also possible
though perhaps less likely ,
a sort of double salt or complex salt of normal lead styphnate
and guanylazide styphnate .
The nitroguanylazide is interesting too , but I have not
found any reference describing its explosive properties ,
except that it is explosive . Whether or not the nitroguanylazide forms stable explosive salts similarly as does the guanylazide is unclear .
Linked below are some other threads which contain information relevant to this thread with regards to precursors and intermediates , particularly of
interest being methods based on OTC materials . This list will be lengthened here and/or further along in this thread as time
passes .
OTC Cyanamide
https://sciencemadness.org/talk/viewthread.php?tid=825
Guanidine
https://sciencemadness.org/talk/viewthread.php?tid=1469
Cyanuric Acid
https://sciencemadness.org/talk/viewthread.php?tid=8160
[Edited on 25-3-2007 by Rosco Bodine]
Attachment: US2125462 Guanylazide Picrate Base Charge.pdf (120kB)
This file has been downloaded 2876 times
Referring to "Green primaries 1"
pdb - 29-3-2007 at 06:39
I am considering preparing some of the primaries listed in the "Green primaries 1" paper available upper in this thread. However, I have two questions
before going further:
- the ferrous complex requires [FeII(H2O)6]Cl2 : I have been searching chem databases for a while, and I didn't find this 6-hydrate chloride, but the
4-hydrate form only. And I don't think it's a typo in the article. If the molar ratio is respected, do you think it could make a difference ?
- is it known if such green primaries are unequivocal ?
Rosco Bodine - 29-3-2007 at 07:58
I could be wrong but I would expect that any soluble Iron
compound could be substituted in these syntheses ,
and also for the copper and ammonium variants . I see no reason why such substitutions would cause any problem .
I would avoid the acetates and other organic acid salts completely if possible
Nitrates would be my first choice .
I believe there is something in the articles which mentioned about a hundredfold variation in the
explosiveness , dependant upon the substituents
for the complex . I'd have to go back and read it again
but I'm sure it is there because I wondered what would be the effect of a potassium substituent on the level of hydration , and couldn't find anything
. I figured it was something that they were looking at also within the
experiments that are no doubt still a work in progress .
[Edited on 29-3-2007 by Rosco Bodine]
The_Davster - 12-6-2007 at 21:42
A previous limitation in the prep of nitrotetrazoles from 5-ATZ was the nature of the acid copper nitrotetrazolate salt, Cu(NT)2HNT,(NT=CN5O2-)
This patent here: http://www.freshpat ents.com/Primary-explosives-dt20060209ptan20060030715.php mentions:
"Ammonium nitrotetrazolate was prepared by diazotization of 5-aminotetrazole in the presence of excess nitrite followed by extraction as the
tri-laurylamine salt and displacement by ammonia. Upon addition of stoichiometric amount of ammonium hydroxide, sodium nitrotetrazolate forms
quantitatively and is analytically pure."
I have seen nothing in the literature on such a synthesis, I wonder if it is some sort of in-house method at LANL or something?
Axt - 12-6-2007 at 22:46
I've tried to find a detailed direct prep. and isolation for the sodium salt before but was met with much frustration as no authors give details,
rather refer it off to the attached article, which refers it off to the unobtainable reference:
Gilligan, W. H., and Kamlet, M. J., Technical Report 76-146 (1976), Naval Surface Weapons Centre, White Oak Labs, Silver Spring, Md, U.S.A.
[Microfiche AD A036 0861.
Some info can be gleaned from the attachment though.
"1 H-Tetrazol-5-amine monohydrate was converted into sodium 5-nitrotetrazolate dihydrate by diazotization in the presence of excess nitrite according
to the method of Gilligan and Kamlet."
"Sodium 5-nitrotetrazolate dihydrate was prepared from tetrazol-5-amine hydrate according to the method of Gilligan and Kamlet. The product was
purified by dissolution in the minimum of boiling acetone followed by filtration and precipitation by addition of an equal volume of hexane. The
solution was allowed to cool and the product filtered under suction. Compound was obtained as irregular, chunky white crystals; ignition temp. 202",
210°, 214° (lit. 202°)."
So, anyway. The action of sodium nitrite on the intermediate diazonium salt. It seems like no copper ion needed.
[Edited on 13-6-2007 by Axt]
Attachment: Studies of stab initiation. Sensitization of lead azide by energetic sensitizers.pdf (633kB)
This file has been downloaded 3043 times
Microtek - 13-6-2007 at 01:05
I converted the copper salt to the sodium salt by dissolving it in warm water (it is quite soluble at elevated temps) and adding a stoichiometric
amount of NaOH soln to pricipitate Cu(OH)2. Then I kept the suspension at 80-90 C until the hydroxide had decomposed to the oxide to facilitate
filtering. Gentle evaporation of the filtrate gave the hydrated sodium nitrotetrazolate.
The_Davster - 13-6-2007 at 05:49
| Quote: |
Originally posted by Axt
The action of sodium nitrite on the intermediate diazonium salt. It seems like no copper ion needed.
|
I find that rather odd, the copper ions are added to prevent detonations from diazotetrazole formed from small droplets of the acidic 5-ATZ solution
on the walls of the reaction vessel from reacting with the NOx given off by the reaction and forming the diazotetrazole in percents greater than 2
which spontaneously explode in solution. I wonder what they use to prevent this?
US patent 4093623 and an older, unimproved version, 2006954, detail the process of producing the copper nitrotetrazolate, and from it the sodium
salt.
from 4093623;
"During initial experimentation with the von Herz procedure by the U.S. Navy, several problems were encountered. First, during the diazotization there
was a continuous series of minor detonations, which while not harmful in themselves, were pschologically disturbing and did on occasion break
glassware. Moreover, there was the possibility that the potentially dangerous (in the dry state) acid copper 5-nitrotetrazole salt would be spilled
over adjacent surfaces as a result of these detonations. Second, upon completion of the diazotization, the acid copper salt was present as a
voluminous gel-like precipitate which required long periods (6 hours or longer) to separate by filtration and to wash free of impurities. This would
seriously hamper scale-up operations where large quantities would have to be processed."
Also, as I found a few days ago, the gel like nature of the copper salt can be reduced by doing the addition of 5-ATZ, acid, and trace copper over 3h
or so, as opposed to 1.5h as detailed in the patent.
Interesting note at being unable to find synthesis refs Axt, I also been finding that there are often voids of information about these types of
compounds.
[Edited on 13-6-2007 by The_Davster]
Nick F - 17-7-2007 at 06:13
I can't remember if I've mentioned this before (and this connection is too slow for me to bother looking), but I have a fair amount of
5-aminotetrazole. I'd be happy to sell/trade some to anyone who's interested...
JohnWW - 17-7-2007 at 08:25
How are you going to be able to legally send such an highly explosive compound through the post? BTW How did you acquire or make the stuff?
Nick F - 17-7-2007 at 10:28
It seems fairly benign to me. If you heat it on a spoon for example it will decompose slightly energetically, but I certainly wouldn`t call it
explosive. The nitrate salt is pretty cool, if you powder it (probably not very safe!) and soak a bit of NG into it then it`s impressive, to say the
least... As for how I acquired it, well, that`s a secret  .
.
The_Davster - 17-7-2007 at 16:42
lol I remember from an old post on roguesci how you got it...you trickster you.
1kg yes?
Aminotetrazole is non explosive, it decomposes non-explosevly at around 205 C IIRC.
I have a hundred or so grams of aminoguanidine, so I am happy, I doubt I will ever need more for the small scale experiments I do.
Nick F - 17-7-2007 at 16:55
Wow, I`d almost forgotten about roguesci! I remember that when I got the stuff I was wondering if I could do a decyclisation to get an azide. But then
I got myself some of that, too. Scamming became almost an addiction at one point!
(2kg )
Engager - 17-8-2007 at 20:44
| Quote: |
Originally posted by The_Davster
A previous limitation in the prep of nitrotetrazoles from 5-ATZ was the nature of the acid copper nitrotetrazolate salt, Cu(NT)2HNT,(NT=CN5O2-)
This patent here: http://www.freshpat ents.com/Primary-explosives-dt20060209ptan20060030715.php mentions:
"Ammonium nitrotetrazolate was prepared by diazotization of 5-aminotetrazole in the presence of excess nitrite followed by extraction as the
tri-laurylamine salt and displacement by ammonia. Upon addition of stoichiometric amount of ammonium hydroxide, sodium nitrotetrazolate forms
quantitatively and is analytically pure."
I have seen nothing in the literature on such a synthesis, I wonder if it is some sort of in-house method at LANL or something?
|
I've made NH4NTZ solution in such method: dissolve Cu salt with excess of Ba(OH)2 in water, boil until CuO settles down, filter it off and measure
weight to calculate amount of nitrotetrazole ion in solution. After this add solution of (NH4)2SO4 (1 mole for 1 mole initial Ba(OH)2), filter
unsoluble BaSO4 and you have NH4 salt solution with some dissolved ammonia, whitch is removed by boiling. Although i'm not isolated NH4 salt i found
that it is very soluble. Also i found that condidions on whitch NH4NTZ and Fe salts are mixed, are important. After addition of FeCl2 and 2h boiling i
got no precepitate. But then i dissolved CoSO4 or CuSO4 in sample taken from solution, i got pink or blue precipitate of corresponding
nitrotetrazolate. On addition of K2CO3 to sample and slight heat i got ammonia smell. So i concluded that solution contains NH4 nitrotetrazolate, so
my reagents are ok. Some special conditions are needed to form NH4FeNT salt. I'm sure in that. Also i found that in patent they use Fe(ClO4)2 instead
of FeCl2 used in green primaries article. Perchlorate anion very unlikely goes to inner sphere of complex compound, so this change may have some
reason, may be to assist complex formation or to minimize side reactions, but i am not sure.
[Edited on 11-9-2007 by Engager]
Engager - 25-8-2007 at 22:48
I have succeded making 5-nitotetrazole - ammonium complex NH4CuNT. Compound formula (NH4)2[Cu[NTZ]4(H2O)2]. I've made it by following method: Solution
of 5.5g ammonium 5-nitrotetrazolate in 38 ml of water added with stiring to solution of 2.52г Cu(NO3)2*6H2O in 110 ml H2O. A small quantity of
blue precipitate is formed emidately. Solution was heated on boiling waterbath for 4 hours, solution becames transparent blue. It's slowly cooled to
room temperature and after to 10C in freezer. Large quantity of blue "snowy" precipitate is formed, solid is filtered off, washed with ice cold water
and with small portion of ice cold alcohol. Product was air dried. Photo of product shown below:
According to patent data, density is 1.94 g/cm3, detonation velocity 7390 m/sec (at 1.71 g/cm3). Insensitive to spark up to 0.36 J (human activity
generates up to 0.25 J), sentive to shock 23 cm (vs 9.6 PbN3 and 14 PETN), slightly sensitive to friction 0.6 kg (vs 0.01 PbN3 and 5.8 for PETN).
Thermicaly stable up to 265C, detonation products volume is about 750 l/kg, products of explosion: N2, CO2, H2O, ~2% NO2, ~3%CO. Oxygen ballance (CO)
is zero. Substance is stable on air, light and moisture. Almost completely safe then wet, even with open flame. In dry state flame contact takes DDT
(deflagration-detonation transition).
I've also tried Fe and Co complexes. Attempt with FeCl2*6H2O was unsuccesfull, as i've mentioned in previous post. Attempt with Co(ClO4)2*6H2O gave
success but yield was low. I guess there are some special conditions that need to be satisfied then making Fe and Co complex 5-nitrotetrazolates.
[Edited on 26-8-2007 by Engager]
Engager - 25-8-2007 at 23:15
| Quote: |
Originally posted by Rosco Bodine
See PATR , Vol.1 , A-569
The sodium salt is produced in 76% yield from a solution of
5-aminotetrazole in 15% NaOH at 50C by oxidation with KMnO4 solution added dropwise . Unreacted KMnO4 is
decomposed with alcohol , and the solution is refluxed for 1 hour at 100C . On cooling the sodium salt is obtained as
crystals . |
Synthesis of Na-5,5'-Azotetrazolate
Method was tested with success. 5-Aminotetrazolate monohydrate (10 g) was dissolved with stirring in 40 ml of 15% aqueous NaOH solution at 50°C
(dissolves amlost permanently, solution is colorless). Another solution of 10 g of KMnO4 in 50 ml of hot distilled water was prepared. The aqueous
KMnO4 solution was then added slowly into the stirred aqueous NaOH solution of 5-aminotetrazolate monohydrate (small gas evolution and heating were
observed). Resulting solution is dark green with some brown precipitate. Into this mixture 10 ml of ethanol was added to react with excess KMnO4,
sollution color is turned to brown/black. Then, the reaction solution was refluxed at 100° C. for 1 h. The resulting reaction mixture was then
filtered. Upon cooling, yellow crystals of sodium 5,5'-azotetralate dihydrate (SZT) crystallized from the filtrate gradually. The crude product was
recrystallized and dried to give 9.13 g (76.4%) of pure SZT. Photo of product crystallizing under mother liquer, and solid product on filter shown
here:
Reaction scheme:
[Edited on 11-9-2007 by Engager]
Engager - 9-9-2007 at 23:47
I've also made two other tetrazole-based energetic compounds. Diazoaminotetrazole and dihydrazinium 5,5'-azotetrazolate (mentioned as substance with
highest known positive heat of formation).
Synthesis of sodium bis-5,5'-diazoamintotetrazolate from aminoguanidine bicarbonate
Prepare mixture of 11.5 ml 70% nitric acid with 100 ml of water, add by portions with stirring 24.8g aminoguanidine bicarbonate. Stir mixture until
CO2 evolution stops and then add 20.4 ml 70% acetic acid. Mixture is stirred and slightly heated until all solid dissolves. The resulting clear
yellow solution is solution of aminoguanidine nitrate in 12-13% acetic acid. This solution is cooled in freezer to 3-4C, well mixed and placed on ice
bath. Slowly, with stirring, by small portions at time ice cold solution of 17.5g sodium nitrite in 75 ml of water is added. While addition,
temperature must be all times kept below 12C, perfectly in interval of 5-7C, process takes about 30-40 minutes. After diazotation is finished mixture
is removed from ice bath and left to stand at room temperature for 24 hours. Some time after removal from ice bath slow evolution of nitrogen begings,
and mixture can heat up to 25C, this is ok, so don't worry, and after about 12-16 hours of standing evolution of nitrogen stops and large amount of
diazoaminotetrazole precipitates. Solid is filtered off, washed with ice cold water slightly acidified with acetic acid and left to dry at room
temperature. Yield is about 50% of pure mono-sodium salt of diazoaminotetrazole. Photos of product shown below:
Reaction scheme:
[Edited on 11-9-2007 by Engager]
franklyn - 10-9-2007 at 07:36
Ooh very nice , this one is certainly worth pursuing. Azotetrazolate is a dianion , its salts can
have two cations or ligands. It too would be interesting to see if a polymer resin can be formed
with formaldehyde.
| Quote: |
Dihydrazinium 5,5’-Azotetrazolate
Synthesis in water produces yellow needles of the dihydrazinium salt [N2H5]2:[N4C-N=N-CN4].
Heat of formation is + 1147 kcal kg one of the highest ever reported. The compound is stable at
room temperature, almost insensitive to friction and impact, but detonates violently when the
explosion is initiated, e.g., by rapid heating over the decomposition temperature or by using an
initiator.
5,5’-azotetrazolate salts show the remarkable insensitivity to electrostatic discharge, friction
and impact while having a very high heat of formation.
|
Dihydrazinium 5,5’-Azotetrazolate Dihydrazinate Complex

http://pubs.acs.org/cgi-bin/abstract.cgi/inocaj/2001/40/i14/...
US patent 5877300
Preparation of guanidinium 5'5-azotetrazolate
Variations on tetrazolates _
Ionic Liquids as Energetic Materials
http://stinet.dtic.mil/cgi-bin/GetTRDoc?AD=ADA464308&Loc...
.
Engager - 10-9-2007 at 08:07
I've already made dihydrazinium azotetrazolate. I can post metod of synthesis with photos if somebody interested. It's yellow needle like solid,
soluble in water.
[Edited on 11-9-2007 by Engager]
Engager - 10-9-2007 at 20:27
Synthesis of dihydrazinium 5,5'-Azotetrazolate (HZT)
Prepare solution of 4.8g sodium 5,5'-azotetrazolate in 30 ml boiling water and 5.58g barium chloride dihydrate in 15 ml of boiling water. Solutions
are mixed and stirred, precipitate of barium 5,5'-azotetrazolate forms emidately, solution is cooled to 10C and filtered. Ba salt is washed with small
amount of ice cold water and dried at room temperature. Yield is about 6.2g.
Make solution of 5.3g hydrazine sulphate (N2H6SO4) in 155 ml of water, 6.44g of barium hydroxide is added with stirring, and after 6.2g of barium
5,5'-azotetrazolate is added. Mixture is well stirred for 1 hour, solid (BaSO4) is filtered off and discarded, resulting in yellow solution of
dihyrazinium 5,5'-azotetrazolate. Solution of HZT is placed on boiling water bath and heated until most of water evaporate and first crystals of HZT
form. Solution is then removed from water bath and cooled to room temperature and after in freezer to 10C. Mixture almost comepletely solidifies to
form yellow needles of dihydrazinium 5,5'-azotetrazolate dihydrate. Crystalls are filtered of and dried at room temperature. Yield is about 87%.
Anhydrous salt may be obtained by drying dihydrate in vacuum exicator at 100C for 2 days.
I've already made dihydrazinium azotetrazolate. I can post metod of synthesis with photos if somebody interested. It's yellow needle like solid,
soluble in water. Photos shown below, left photo is barium azotetrazolate, two others are dihydrazinium azotetrazolete (HZT):



Reaction scheme:
[Edited on 14-9-2007 by Engager]
The_Davster - 10-9-2007 at 21:13
| Quote: |
Originally posted by Engager
Synthesis of sodium bis-5,5'-diazoamintotetrazolate from aminoguanidine bicarbonate
|
What is the referance for this procedure? I have not came across it, and I have read much of the azotetrazolate literature.
| Quote: |
Originally posted by franklyn
It too would be interesting to see if a polymer resin can be formed
with formaldehyde.
|
If the chemistry is analogous to that of nitrotetrazole, an alcohol will be formed, in the case of nitrotetrazole NO2CN4CH2OH is formed
[Edited on 10-9-2007 by The_Davster]
Engager - 10-9-2007 at 21:45
Synthesis of 5-Aminotetrazole (ATZ)
Thiele method. 34g aminoguanidine-bicarbonate is dissolved in 217 ml of 15% nitric acid (36ml of
70% HNO3 + 185 ml water). Mixture is stirred until CO2 evolution stops and all solid dissolved. Resulting solution is diazotized by solution of 17.2g
sodium nitrite in 35 ml of water. Nitrite solution is added slowly with stirring, while reaction mixture is cooled on ice bath, temperature must all
times kept below 20-25C. Diazotation is proceeding smothly with neglible evolution of NOx, if mixture is foaming (result of HNO2 decomposition),
addition must be paused and mixture must be well stirred to stop foaming, before new portions of nitrite added. Addition takes time about 10-15
minutes. After addition of nitrite is completed, mixture is allowed to sit for 20 mins at room temperature, and 29g of Na2CO3 (or 46g of baking soda)
is added by portions with mixing. Mixture is stirred untill CO2 evolution stops and all excess of bicarbonate fully dissolves. Mixture is placed to
round bottom flask with attached reflux condenser and boiled for 4 hours. Resulting solution of 5-aminotetrazole is acidified by 30% H2SO4 to pH=4,
and left to cool to room temperature. Usualy crystallization of product starts around 40C, but solution have great tendency to supersaturate. If after
cooling to room temperature crystallization is not started, seed crystall of aminotetrazole is introduced (made by placing glass rod with drop of
solution to alcohol), or inner side of flask (below solution of course) is rubbed with glass rod with intense friction. After crystallization is
started solution is left at room temperature for 12 hours, and cooled to 10C in freezez. Crystalls of 5-aminotetrazole are filtered, slightly washed
with ice cold water and dried at room temperature. Yield is 13.6g (64%). Photo of product will be shown at bottom of post.
Reaction scheme:

Schtolle method. Warning!!! This method includes work with
exteremely dangerous, explosive and highly toxic hydrogen azide. Concenration of it's solution must be kept below 20% all the times because of severe
explosion hazard (<20% are explosion safe). All work must be done with good ventilation, and fumes must not be inhaled in any circumstances (HN3 is
very volatile, and is effective protoplasmic poison, causes blood cells decomposition and severe headaches). Never add sodium azide to cold acids
solutions - it may result in condensation of drops rel. conc. HN3 on cold walls of flask, and can explode with extreme violence. Method of synthesis
is optimized for maximum safety, but precautions must be remembered all times. Dissolve 10.5g of dicyandiamide and 16.25g of sodium azide in 250 ml of
water heated to 50C. Mixture is stirred untill all solid dissolve, and 2.15 ml of 36% HCl is added dropwise with stirring, mixture is left at room
temperature for 12 hours, then, after heating to 50C 4.3 ml of 36% HCl is added in same way, mixture is left for 6 hours, next 6.45 ml and 2-3 hours,
and finaly remaining 8.6 ml of 36% HCl. After addition is completed mixture is left standing at room temperature for 1 week, white crystalls of
5-aminotetrazole must apper, but solution again may be supersaturated and no crystalls are precipitated - in this case crystallization must be started
as written in Thiele method above. Yield depends of standing time and puriry of reagents, i have ~50% (10.5g) yield after 2 weeks of standing, but
literature sources show yeilds up to 97% after longer standing. Pure snow white crystalls of 5-aminotetrazole are filtered off, washed with ice cold
water and dried at room temperature.
Reaction scheme:

Photos of products are shown below. Left photo is ATZ made by Thiele's method, and right one is photo of ATZ made by
Schtolle's method:


[Edited on 11-9-2007 by Engager]
Engager - 10-9-2007 at 22:02
| Quote: |
Originally posted by The_Davster
What is the referance for this procedure? I have not came across it, and I have read much of the azotetrazolate literature. |
Reference is russian book: Бубнов П.Ф.
Инициирующие
взрывчатые вещества и
средства инициирования
(часть 1). М., 1940. [P.F.Bubnov "Primary explosives and prime devices(part 1)" Moscow, 1940.] page 316
Original method uses aminoguanidine nitrate as starting material, i have modified it to generate it in situ from aminoguanidine bicarbonate. Method
was tested by me with success, photo is evidence if you don't believe. Also i want to post reaction mechanism so no further explanation will be
requiered:
[Edited on 11-9-2007 by Engager]
Engager - 10-9-2007 at 22:54
Somebody interested in synthesis procedures for BNCP [Tetraamino-cis-bis(5-nitro-2H-tetrazolato-N2) cobalt (III) perchlorate] and NH4CuNT
[5-nitro-1H-tetrazolato-N2 cuprate (II)], both newest edge priming explosive materials?
[Edited on 11-9-2007 by Engager]
artem - 12-9-2007 at 06:22
| Quote: |
[quote/]
..dihydrazinium salt [N2H5]2:[N4C-N=N-CN4]. Heat of formation is + 1147 kcal kg one of the highest ever reported... |
Calculated value H=+1147kcal/kg (1105kJ/mole) is doubtful. Different estimations give no more than +715...750KJ/mole (743...779kcal/kg).
For example,
condensed polyacetylenes 2055kcal/kg(C4H2), 1570(C4N2)
azides 1467(HN3), ~1490(C(N3)4), ~1400 for C2N4(N3)2, N3CN4H, 1100-1200(C3N3(N3)3).
tetrazole 817
Rosco Bodine - 13-9-2007 at 15:41
It's nice to see interest in experimentation with these tetrazoles Nice
crystals
Some thoughts ...
There are a few related things which
may be worth looking into , which were mentioned on the preceding page .
There are possible shortcuts to aminoguanidine from reaction of any guanidine salt with hydrazine via hydrazine sulfate . See US5041661 . And also
possibly dicyandiamide reaction with hydrazine sulfate may produce aminoguanidine . See US3285958 .
Guanylazide styphnate , and nitroguanylazide styphnate could also be very interesting
As styphnic acid is a di-acid , the neutral salts would actually be (di)guanylazide styphnate , and likewise for the (di)nitroguanylazide styphnate
salt . There probably would be a basic lead salt for the mono-guanylazide styphnate and also for mono-nitroguanylazide styphnate .
Also the synthesis of aminotetrazole might produce higher yield via tetracene intermediate .
[Edited on 14-9-2007 by Rosco Bodine]
Engager - 14-9-2007 at 03:37
Synthesis of acid copper 5-Nitrotetrazolate (CuNT) from 5-aminotetrazole
Prepare solution 20.8g sodium nitrite and 11g of copper sulphate (CuSO4*5H2O) in 60 ml of hot water, resulting
solution contains copper nitrite and has dark green color. Prepare solution of 10.3g aminotetrazole (ATZ) and 0.4g CuSO4*5H2O in 12.8 ml 70% nitric
acid + 120 ml of water. Diazotetrazole is intermediate in nitrotetrazole synthesis, it can explode in solution if concentration will reach 2% from the
slightless stimulus, even at 0C. Microexplosions are not dangerous but acting on nerves. To completely avoid them, slow addition, effective stiring
and carefull temperature control are essential. Small adition of copper sulphate to ATZ solution is essential to avoid microexplosions in drops, on
contact with nitrogen oxides, escaping from reaction mixture. Copper nitrite solution is placed on ice bath and cooled to 5C, then solution of ATZ in
nitric acid, is added slowly with stirring (perfectly drop by drop). During addition temperature of reaction mixture must be kept below 15C all times.
If mixture begins to foam, addition is paused and mixture is well stirred until foam dissapears before next portions of ATZ solution are added
(foaming is result of HNO2 decomposition to water and NOx if it's concentration is too high). Reaction is proceeding smoothly, without
microexplosions, with small evolution of NOx, if conditions are carefully controlled. Whole addition process takes time about 1.5 hours. Close to end
of addition mixture becomes thicker, and changes color to green- blue (similar with homemade CuCO3*Cu(OH)2). After addition is completed mixture is
left to sit in ice bath for 15 minutes, after that 14 ml of 70% nitric acid + 6 ml of water is added with stirring. Reaction mixture is left for 1
hour, solid precipitate of acid copper salt of nitrotetrazole is filtered off, washed with 5.72 ml HNO3 + 44 ml H2O and with three portions of 50 ml
H2O. Yield is about 85%. Product is bluish - green crystals, almost insoluble in cold water.
Warning!!! Acid copper salt of nitrotetrazole is powerfull and sensitive explosive.
It is almost completely safe then wet, but in dry state it can explode violently on friction, impact or heating. Safety precautions must be
remembered all times.
Below are the photos of process. Left photo shows solutions of copper nitrite (left one) and ATZ in nitric acid with
CuSO4 added (right one). Photo in the middle shows ice bath with sitting reaction flask, and termocouple temperature control. Right photo shows
reaction product on filter.
Reaction sheme:
Acid copper salt of nitrotetrazole can be easily converted to soluble salts of nitrotetrazole by heating in solution
of corresponding hydroxides. For example solution of sodium 5-nitrotetrazolate can be prepared by boiling acid copper salt in NaOH solution:
Cu(NT)2*HNT + 3NaOH => 3NaNT + CuO+ 2H2O. Solid black copper oxide is removed by filtering, pure solution of NaNT can be concentrated to separate
solid salt, or can be used dirrectly for further reactions.
[Edited on 14-9-2007 by Engager]
Axt - 28-9-2007 at 03:29
Excellent posts Engager, but just a suggestion, dont use imageshack or other free hosts as inevitably within a month or two they will all end up dead
links. Instead either attach them directly of to imbed then in the text use the <a
href="http://www.sciencemadness.org/talk/viewthread.php?tid=603">forum pic ftp</a> setup specifically for this purpose. What program did you
use to draw the reaction schematics?
I recently prepared 5-nitraminotetrazole. Its properties given in Chem. Mater. 2007, 19, 1731-1739 are quite remarkable, its density via gas
pycnometry is 2.06g/cm which gives a calculated VOD of 10358m/s using the "Cheetah 4.0" program of LLNL. JACS, 73(5), 2327-2329 mentions that is a
sensitive explosive, though this is disputed by J. Org. Chem., 18(8), 941-945, saying that it wont explode when struck "very sharply" on an anvil with
a hammer. Though they dont say if they were testing the sensitivity of the hydrated or anhydrous acid. Thats another problem with nitraminotetrazole,
it retains solvent of crystalisation and water of hydration though this is said to be lost of standing at room temperature.
I prepared nitroaminoguanidine by condensing <a href="http://www.sciencemadness.org/talk/viewthread.php?tid=8911">nitroguanidine</a> with
hydrazine hydrate via the method of JACS, 73, 474, this is said by the authors to give a purer product in higher yield then the method of JACS, 50,
2465-2470 that used hydrazine sulphate-ammonia mixture of for the condensation. Below is the change in colour of the solution and the appearance of
nitroaminoguanidine crystals under the microscope.
<center><img src="http://www.sciencemadness.org/scipics/axt/nitroaminoguanidine-crystals.jpg"></center>
To see if this was indeed nitroaminoguanidine, the hydrazone derivative was produced by condensation with formaldehyde. On standing very long fine
needles separated which meets the description in JACS, 50, 2465-2470. The precipitate resembled fiberglass or nitrocellulose and when ignited
deflagrated vigourously with a large orange flame as shown below.
<center><img src="http://www.sciencemadness.org/scipics/axt/nitroaminoguanidine-formaldehyde-deflag.jpg"></center>
The nitrosation of nitroaminoguanidine was done following the experimental method "A" given in JACS, 73, 2327-2329, which uses a heated neutral
solution of nitroaminoguanidine and potassium nitrite, though modified it to use sodium nitrite. Cooling the solution resulted in precipitation of the
sodium nitraminotetrazolate but this was not isolated, rather HCl was added then it was extracted with ether, evaporation yielded ~5.5g of crude
5-nitraminotetrazole from 8g nitroaminoguanidine. Doing this probably resulted in some contamination with nitroguanylazide and its this contamination
that J. Org. Chem., 18(8), 941-945 speculates to be the reason form an increased sensitivity. This crude product on drying for a number of days at
room temperature exploded easily under the hammer on a rusty anvil and deflagrated in a flash on ignition as shown in the picture below.
<center><img src="http://www.sciencemadness.org/scipics/axt/nitraminotetrazole-deflag.jpg"></center>
A 1:1 molar aqueous mixture of AgNO3 and nitraminotetrazole were combined which resulted in an immediate white precipitate of the acid silver salt.
When dried this salt only "snappled" when held in a flame spreading the pile around but wouldnt sustain in the small quantities used.The literature
does mention that salts of nitroaminotetrazole are not initiating explosives with the possible exception of the mercury salt. The potassium salt was
also prepared by combining 1:2 molar ratio of nitraminotetrazole and potassium hydroxide in ethanol, again a white precipitate formed immediately
which was filtered and dried. This only deflagrated on ignition as shown below.
<center><img src="http://www.sciencemadness.org/scipics/axt/potassium-nitraminotetrazole.jpg"></center>
[Edited on 1-10-2007 by Axt]
Engager - 30-9-2007 at 15:13
| Quote: |
Originally posted by Axt
Excellent posts Engager, but just a suggestion, dont use imageshack or other free hosts as inevitably within a month or two they will all end up dead
links. Instead either attach them directly of to imbed then in the text use the <a
href="http://www.sciencemadness.org/talk/viewthread.php?tid=603">forum pic ftp</a> setup specifically for this purpose. What program did you
use to draw the reaction schematics?
[Edited on 28-9-2007 by Axt] |
ChemDraw Ultra 8.0 from ChemOffice 2004 Suite. Thanks for the advice, next time i will attach images dirrectly to posts. Now, getting back to topic.
I've recently made silver salt of 5-nitrotetrazole, and conducted some tests on AgNTZ senivity. Here is a small detail about this interesting
explosive material:
Silver nitrotetrazolate is white crystaline powder, stable to light and moisture, prepared by mixing soluble nitrotetrazole salt solution with silver
nitrate. Compound is powerfull primary explosive, heat of explosion 1.94 MJ/kg, initiating power is 0.005g vs tetryl (HgNTZ: 0.006g, Lead azide:
0.02g, Mercury Fulminate 0.2g). Silver 5-nitroterazolate is sensitive to shock and friction - slighly more then mercury fulminate.
Can be overpressed.
Then wet it can detonate on contact with flame, but is much less sensitive to friction and shock, then in dry state. I've made some friction and tests
on dry salt. On intensive friction between paper and wooden rood, no explosion was encountered. However attempt of grinding in mortar, resulted in
almost emidate violent explosion, with bright white flash. Sensivity to shock is very high, even slightest hits can cause violent detonation. In my
personal oppinion, silver nitrotetrazolate can be handeled in safe manner with little risk when threated right, but extreme caution is advised.
I've attached photo of silver 5-nitrotetrazolate, among other tetrazole derivatives i've made so far. Upper-left to lower-right: 1. Copper-ammonium
complex 5-nitrotetrazolate (NH4)2[Cu(N4C-NO2)4(H2O)2] - green primary (NH4CuNT) ; 2. (N2H4)2(N4C-N=N-CN4) - dihydrazinium 5,5'-azotetrazolate (HZT) ;
3. Cu3(N4C-N=N-NH-CN4)2 - copper 5,5' diazoaminotetrazolate (CuDAT) ; 4. Cu(N4C-NO2)2*(HN4C-NO2) - acid copper salt of 5-nitrotetrazole (CuNT) ; 5.
Ag(N4C-NO2) - silver 5-nitrotetrazolate (AgNT) ; 6. Na3(N4C-N=N-NH-CN4) - sodium 5,5'-diazoaminotetrazolate (NaDAT) ; 7. Na2(N4C-N=N-CN4) - sodium
5,5'-azotetrazolate (NaAT) ; 8. HN4C-NH2*H2O - 5-aminotetrazole monohydrate (ATZ).
[Edited on 1-10-2007 by Engager]

The_Davster - 30-9-2007 at 15:45
Since I was feeling left out in my pet thread...
I have been too busy to experiment!
But when I have time, time for these guys, among other tetrazoles.
1) 2-hydroxylmethyl-5-nitrotetrazole
6.85g of sodium nitrotetrazole (hydrate) was dissolved in 50mL of water and 24.5mL of 20% sulfuric acid and 10mL of a 37% aqueous solution of
formaldehyde were added with stirring at 5-10 C. The mixture was kept for 24h at 18-20C and extraced with ether(3x50mL) and the extract dried over
magnesium sulfate and evaporated under reduced pressure to obtain 3.2g(67%) of compound 1 as a colorless crystalline substance, mp 63-64C
2) 2.6g of (1) was dissolved in 8mL of 101% sulfuric acid at 18-20C and 0.27g of paraform was added. The mixture was stirred for 1h at 35-40C, cooled
to 5C, and poured into 50g of finely crushed ice. The precipitate was filtered off and washed with 50mL of ice water. Yield 4g(80%), colorless
crystalline substance, mp157-158C
3)1.45g of (1) was dissolved in 20 mL of 101% sulfuric acid at 18-20C and the mixture was stirred for 2h at that temperature and poured into 100g of
finely crushed ice. The precipitate was filtered off and washed with 50mL of ice water. Yield 0.5g(35%), colourless crystalline substance, mp
165-167C
[Edited on 30-9-2007 by The_Davster]

Engager - 7-10-2007 at 16:57
I've found a very interesting nitrotetrazole based compound in my russian explosive related documents, called HT-1:
1,3-di(nitrotetrazolato-N2)-2-nitro-2-azopropane. Structual formula computed by RHF is attached below this message. HT-1 can be made by reaction of
1,3-dichloro-2-nitro-2-azopropane with silver nitrotetrazolate in organic solvent:
Cl-CH2-N(NO2)-CH2-Cl + 2Ag(N4C-NO2) => (N4C-NO2)-CH2-N(NO2)-CH2-(N4C-NO2) + 2AgCl (precipitate)
Some properties of HT-1. First time prepared in USSR in 1972, melting point 165C, density 1,90 g/cm3, heat of formation +476 ccal/kg (according
different source 388 ccal/kg), heat of explosion 1430 ccal/kg, detonation velocity (density): 9350 m/sec (1.86), 9940 m/sec (1.90), critical diameter
0.82 mm. Thermal stability is acceptable.
I have fair ammount of Ag nitrotetrazolate, all that i need is to synhesis of 1,3-dichloro-2-nitro-2-azopropane. I had an idea to make this stuff
through the nitramide, whitch i've made by hydrolisis of N,N'-dinitrourea in water (watch dinitrourea thread). Wonder if this will work:
NH2-NO2 + 2CH2Cl2 => (ClCH2)2N-NO2 + 2HCl
Other ideas:
2CH2Cl2 + NH2SO3K (potassium sulfamate) => (ClCH2)2NSO3K + 2HCl => (ClCH2)2NSO3K + HNO3 => (ClCH2)2N-NO2 + KHSO4
(CH3)2NCOH(DMF) + HNO3 => (CH3)2N-NO2 + CO + H2O => (CH3)2N-NO2 + Cl2 => (ClCH2)2N-NO2 + 2HCl
Someone have any ideas about how 1,3-dichloro-2-nitro-2-azopropane can be made?
[Edited on 8-10-2007 by Engager]

Engager - 7-10-2007 at 17:15
There is also an interesting route from 5,5'-azotetrazole (which is made by simple oxidation of aminotetrazole by KMnO4; watch my post somethere
above) to isocyanogen tetrazide (watch US patent 2,990,412 for method details). Compound can be easily made from isocyanogen tetrabromide and solution
of NaN3 in acetone/water. Trick is that isocyanogen tetrobromide can be simply made with good yield by action of bromine (in form of bromine water) on
solution of sodium azotetrazolate, nitrogen is evolved and product separates as black oily layer:
Na(N4C)-N=N-(CN4)Na + 3Br2 => 2NaBr + 4N2 + Br2=C=N-N-C=Br2
Action of sodium azide in acetone/water gives isocyanogen tetrazide with excelent yield:
Br2=C=N-N-C=Br2 + 4NaN3 => (N3)2C=N-N-C(N3)2 + 4NaBr
Not much digital data about explosive properties of compound exists, but it is certainly powerfull explosive. Structual formula calculated by RHF is
attached below this post. May be someone want to try this? This stuff is 100% real and excelenly described in original work of Thiele and in US
pattent i've mentioned. Unfortunately i can't try this myself due to the lack of good place for such experementation. Someone interested in trying
this stuff?

The_Davster - 7-10-2007 at 18:22
Engager, your posts in this thread are always a pleasure!
HT-1 is very similar to another energetic:

The preparation of which is in the attachment.
I know I came across the synthesis of 1,3-dichloro-2-nitro-2-azapropane while looking for the synthesis of 1-chloro-2-nitro-2-azapropane, I will use
scifinder at Uni on tuesday to look it up again.
Attachment: 1nitrotetrazolato2nitro2azapropane.pdf (113kB)
This file has been downloaded 2487 times
Engager - 8-10-2007 at 06:28
| Quote: |
Originally posted by The_Davster
HT-1 is very similar to another energetic. |
Yes i know this one, i asked for this article in References, section. Thiele work with isocyanogen is also avialable there by my request. I have
description of manufacture 1,3-dichloro-2-nitro-2-azoprapane from US patent 4,085,123 witch is attached to the message. But unfortunately i haven't
got acetic anhydride and high vacuum apparatus to preform such preparation. What's why i'm looking for simplier ways...
[Edited on 8-10-2007 by Engager]

franklyn - 8-10-2007 at 17:14
| Quote: |
Originally posted by Engager
Isocyanogen tetrazide easily made from isocyanogen tetrabromide and solution of NaN3 in acetone/water |
2,4,6-trichlorotriazine ( Cyanuric trichloride ) similarly serves as the precursor for
2,4,6-triazidotriazine , also made by metathetic action with NaN3. ( See page 432
Chemistry of Powder & Explosives by T. L. Davis.
2,4,6-triazidoborazine , sensitive to impact and friction , one of the very few known
metastable boron compounds ( all azides ) is obtained from a 1 : 3 molar blend of
trichloroborazine and trimethylsilylazide ( Me3SiN3 )
http://www.sciencemadness.org/talk/viewthread.php?tid=1987&a...
the resulting trimethylsilylchloride evaporates from the product.
.
[Edited on 14-2-2008 by franklyn]
Engager - 30-10-2007 at 02:22
Here is synthesis of 2 copper-nitrotetrazole complexes, both are efficent primaries, and use soluble nitrotetrazole
salts as precursors. This salts can easily be made dissolving acid cooper nitrotetrazole salt (made as described in posts above)in corresponding
hydrohide, heating the solution and filtering off copper oxide. By weighting copper oxide, quantity of nitrotetrazole anion in solution is determined,
this reqired to be known before any other transformations.
Synthesis of precursor nitrotetrazole salts:
Sodium nitrotetrazolate required for first complex is obtained by dissolving acid copper nitrotetrazolate in NaOH
solution. Ammonium nitrotetrazolate is somethat more difficult to produce because ammonium hyrdoxide can not be used dirrectly (copper forms complexes
with it). So to make ammonium tetrazolate acid copper nitrotetrazole salt is dissolved in barium hydroxide, solution is heated to complete exchange
and destroy Cu(OH)2, black copper oxide is filtered off, and weighted to determine quantity of nitrotetrazolate ion in solution. Then ammonium
sulphate is added in stechiometrical ammount to initial barium hydroxide, this results in formation of ammonium nitroterazolate and insoluble barium
sulphate, while excess of barium hydroxide is also destroyed to form some ammonia and barrium sulphate. Solution is filtered from BaSO4 and boiled to
remove residual ammonia, resulting in pure NH4NT solution.
Synthesis of mono-(5-nitro-H-tetrazolato-N)triammine copper(II) perchlorate (MNCuP):
Prepare solutions 10g copper nitrate trihydrate (0.041 mol) in 50 ml of warm water, and 24g (0.25 mol) ammonium
carbonate in 24 ml of 25% ammonia + 30 ml water. Solution of ammonium carbonate is added with stirring to solution of copper nitrate, resulting in
intense blue/violet solution of copper-ammonia complex. Solution is placed on water bath and heated until thin light blue layer of copper basic
carbonate (Cu(OH)2*CuCO3) begin to form at the bottom of reaction flask. During heating period 5g more ammonium carbonate is added in installments.
Solution is filtered from any solid and then 3-4 volumes of alcohol is added. Complex compound is almost insoluble in alcohol and precipitates, solid
is filtered off, washed with alcohol and dried. Product is blue/violet crystalls of [Cu(NH3)3*NH4CO3](NO3). Yield is 75-80%.
To solution of 40 ml 70% HClO4 in 100 ml of water, 3.5g (0.01379 mol) of carbonato triammine copper(II) nitrate in
30 ml of water is added with stirring. Deep blue color of copper carbonate complex quickly turns to transparent sky blue. Resulted mixture is heated
to 60 ± 10С and left to stand at this temperature for half of hour and heated to ~80C, then concentrated solution of sodium 5-nitrotetrazolate
(0.00689 mol) is added by drops with stirring. Mixture is left to sit in boiling water bath for 4 hours, and cooled to room temperature. Product
precipitates as sky blue solid, it is filtered off, washed with small amount of ice cold water and dried. Yield is about 85% of theory.
Photos of products are shown below, left one is carbonato triammine copper(II) nitrate ; right one mono-(5-nitro-H-tetrazolato-N)triamminecopper(II)
perchlorate (MNCuP):
Reaction scheme:
Synthesis of ammonium diaquatetrakis (5-nitro-1H-tetrazolato-N2) cuprate (II) (NH4CuNT):
Solution of 5.5g ammonium 5-nitrotetrazolate (made by chain: CuNT => BaNT => NH4NT) in 38 ml of water added with stiring to solution of 2.52g
Cu(NO3)2*6H2O in 110 ml of water. A small quantity of blue precipitate is formed emidately. Solution was heated on boiling waterbath for 4 hours,
solution becames transparent blue. It's slowly cooled to room temperature and after to 10C in freezer. Large quantity of blue "snowy" precipitate is
formed, solid is filtered off, washed with ice cold water and with small portion of ice cold alcohol. Product was air dried.
Photo of product shown below:
Reaction scheme:
Properties of complexes:
MNCuP: [Cu(NH3)3(NT)](ClO4) - sky blue solid with bulk denstity about 0.65, irregulare plate structure. Sensitive to impact and friction but less then
conventional primaries (similar to BNCP). Digital data about explosive properties is unavialable now, and will be investigated by later research.
NH4CuNT: (NH4)2[Cu(NT)4(H2O)2] According to patent data, density is 1.94 g/cm3, detonation velocity 7390 m/sec (at
1.71 g/cm3). Insensitive to spark up to 0.36 J (human activity generates up to 0.25 J), sentive to shock 23 cm (vs 9.6 PbN3 and 14 PETN), slightly
sensitive to friction 0.6 kg (vs 0.01 PbN3 and 5.8 for PETN). Thermicaly stable up to 265C, detonation products volume is about 750 l/kg, products of
explosion: N2, CO2, H2O, ~2% NO2, ~3%CO. Oxygen ballance (CO) is zero. Substance is stable on air, light and moisture. Almost completely safe then
wet, even with open flame. In dry state flame contact takes DDT (deflagration-detonation transition).
I've also tried Fe and Co complexes. Attempt with FeCl2*6H2O was unsuccesfull, as i've mentioned in previous post. Attempt with Co(ClO4)2*6H2O gave
success but yield was low. I guess there are some special conditions that need to be satisfied then making Fe and Co complex 5-nitrotetrazolates.
[Edited on 30-10-2007 by Engager]
guanylazide 5,5'-Azotetrazolate (possible?)
Rosco Bodine - 30-10-2007 at 12:11
| Quote: |
Originally posted by Engager
Synthesis of dihydrazinium 5,5'-Azotetrazolate (HZT)
Prepare solution of 4.8g sodium 5,5'-azotetrazolate in 30 ml boiling water and 5.58g barium chloride dihydrate in 15 ml of boiling water. Solutions
are mixed and stirred, precipitate of barium 5,5'-azotetrazolate forms emidately, solution is cooled to 10C and filtered. Ba salt is washed with small
amount of ice cold water and dried at room temperature. Yield is about 6.2g.
Make solution of 5.3g hydrazine sulphate (N2H6SO4) in 155 ml of water, 6.44g of barium hydroxide is added with stirring, and after 6.2g of barium
5,5'-azotetrazolate is added. Mixture is well stirred for 1 hour, solid (BaSO4) is filtered off and discarded, resulting in yellow solution of
dihyrazinium 5,5'-azotetrazolate. Solution of HZT is placed on boiling water bath and heated until most of water evaporate and first crystals of HZT
form. Solution is then removed from water bath and cooled to room temperature and after in freezer to 10C. Mixture almost comepletely solidifies to
form yellow needles of dihydrazinium 5,5'-azotetrazolate dihydrate. Crystalls are filtered of and dried at room temperature. Yield is about 87%.
Anhydrous salt may be obtained by drying dihydrate in vacuum exicator at 100C for 2 days.
I've already made dihydrazinium azotetrazolate. I can post metod of synthesis with photos if somebody interested. It's yellow needle like solid,
soluble in water. Photos shown below, left photo is barium azotetrazolate, two others are dihydrazinium azotetrazolete (HZT):
Reaction scheme:
[Edited on 14-9-2007 by Engager] |
(reaction scheme stored on local server)
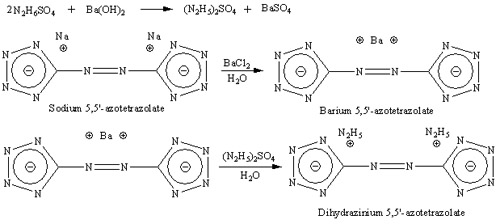
Sorry for not mentioning this earlier ....
It would seem that the guanylazide salt would have more interest in terms of an energetic material and could be made
by a parallel reaction , using (mono)guanylazide sulfate (exist?)
substituted for the dihydrazine sulfate , the parallel
reaction expected to follow the course of your last reaction shown above .
The predicted product would be
guanylazide 5,5'-Azotetrazolate .
The possible usefulness of guanylazide ,
and (though less convenient) nitroguanylazide ,
as well as triaminoguanidine , should not be overlooked
as possible "metal substitutes" in the formation of energetic salts derived from the various energetic acid tetrazoles .
I don't know of any references concerning such materials ,
so extreme caution would be prudent in experiments concerning such possible compounds .
P.S. As Axt pointed out earlier , you should upload , copy your attachments into the forum ftp , so they will not become dead links and empty space ,
(lost data) later , but are safely archived and backed up here .
[Edited on 30-10-2007 by Rosco Bodine]
quicksilver - 12-1-2008 at 14:34
| Quote: |
Originally posted by Rosco Bodine
Yeah it is in PATR under " Tetrazene " , an Australian
government study found that stab detonators containing
tetracene would fail to function after storage at elevated temperatures because of the tetracene slowly changing to
5-aminotetrazole by thermal decomposition . They stated
complete conversion at 90C in less than 3 days .
|
Does anyone know how the decomposition mechanism works within this chemical? I have seen this personally in that after a period of weeks the tetrazene
produced via the COPAE would no longer produce the black-smoke deflagation it had previously. But in this case the exposure level was perhaps 40 C
over the course of weeks or months. Is tetrazene unstable @ room temp?
An addendum: it appears that 40C is not a contributor to instability but 30+ degree temp fluctuation may be. The only accountable issue would be that
or UV from a extremely low level source.
[Edited on 14-2-2008 by quicksilver]
The_Davster - 8-2-2008 at 18:01
synthesis of azidotetrazole from cyanogen bromide:
(as requested)
Attachment: cyanogen azide.pdf (640kB)
This file has been downloaded 2243 times
Engager - 20-2-2008 at 20:44
Today i've made batch of acid copper nitrotetrazolate for fourth time. Unlike my previous experieces, this time i have encountered "minor detonations"
during same procedure as i've used before. These microdetonations are caused by nitrogen oxide fumes from the reaction solution reacting with
droplets of 5-AT on surfaces in the reactor to form 5-diazotetrazole which may spontaneously detonate in
solution when the concentration exceeds 1%. Aside from being psychologically disturbing, these micro-detonations may be strong enough
to break glass and may result in release of the potentially explosive reaction mixture. Detonations are accompanied by white flashes and loud report,
you got to have a steel nerves to stay not worried about such scary stuff, especialy then mixture is allmost full of precipitated acid copper
5-nitrotetrazole salt.
Reasons why this time they arrisen remain unclear, they seem to appear randomly, without any correlation with reaction state. However i've found that
in round bottom flask, they appear much more frequently, then in flat bottom flask, and also depend from flask volume and neck width. This may be
connected with temperature gradients and vent efficency inside reaction wessel.
Temperature also plays a role, surprisingly, when it is low microdetonation probability rises greatly, so it is very important to keep temperature
between 15 and 18C at ALL points of rection mixture. This may be acchived by adjusting reagent additon rate and by effective stirring. Mixing rod edge
must be round and smooth to produce minimal impact if it accidentaly hits the bottom during stirring. At low temperature, glass stiring rod with
relatively sharp edge, causes minor detonations at almost any accidental contact with flask bottom.
So there are some additional notes to procedure to avoid minor detonation problem. Reaction wessel must be flat bottom flask with large volume and
wide neck, stirring rod should not have sharp or low area bottom edge to maximize impact area. Additon rate must be slower at least twice then 90 mins
mentioned in literature, addition shoud be not more then 0.5 ml at one time, perfectly drop by drop. Efficent stirring and temperature control are
essential, temperature during addition time must be kept strictly between 15 and 18C in all points of reacion mixture.
Don't bother literature souces say that minor detonations are relatively safe, whey must be taken very serious!!! They still can do nasty dammage to
glassware, and may be even able to initiate exposion of whole mixture. This synthesis of 5-nitotetrazole must be taken as hazardly dangerous, and must be carried out only by experienced chemist, with all necessary safety measures.
[Edited on 21-2-2008 by Engager]
The_Davster - 22-2-2008 at 22:20
Interesting to hear of your detonations, I have prepared the copper salt in small round bottom or erlenmeyer flasks with magnetic stirring 3 times
now, and have never experianced this. Good to know it can happen even with copper present in the ATZ solution being added. I believe due to a too
slow addition I went to 10C once without experiancing these microdetonations.
Back to info on the tetrazylazide as discussed on the first page of this thread:

Engager - 7-3-2008 at 22:02
Russian references as i promised, sorry man, too large to attach, i had to place them on Rapidshare.
http://rapidshare.com/files/97898324/Davster.rar.html
oket - 8-3-2008 at 04:19
"SOME REACTIONS OF THE AZOTETRAZOLE ANION
WITH DILUTE MINERAL ACIDS"
5-Hydrasinotetrazole
To a solution of disodium asotestrazole pentahdrate (10.0 g) Tuspended in
water (100 ml) was added hydrochloric acid (25 m1 5N). The solution was
warmed on a water bath until the gas evolution finished, then was evaporated
to dryness from water three times to remove the hydrochloric acid. The
residue was dissolved in a minimum of hot water and a hot solution of sodium
acetate (10.0 g) in water (10 ml) was added. The apparatus was flushed out
with carbon dioxide and the solution allowed to cool to give 5-hydrasinotetrasole
as white prisms with a yield of 2.5 g# mp 195 - 8°0 (lit 199d).
Attachment: SOME REACTIONS OF.pdf (1.3MB)
This file has been downloaded 1565 times
Engager - 10-3-2008 at 20:10
Bis-(5-nitrotetrazolato-N2) cobalt (III) perchlorate (BNCP).
Orange-red needle-shaped crystalls insoluble in cold water. Density 2.05 g/cm3, thermicaly stable below 200C, decomposes at 269C. Heat of explosion
3.32 MJ/kg, detonation velocity 8100 m/sec (at 1.97 g/cm3), deflagration - detonation transition takes about 10-11 microseconds, minimal intiating
charge vs RDX is 0.05g. Non toxic, not hygroscopic, sensitive to fire, relatively insensitive to impact and friction (on secondary explosive level).
Gives 50% explosions in drop test with 2.5 kg weight and 17 cm drop altitude (PETN - 12 cm), insensitive to electrostatic discharge. Ignite from Nd
laser beam (wavelenth 1.06 micrometers, impulse time 1 msec, beam diameter 0.5 mm, impulse energy ~ 1.5J). Can be used as primary in detonators
without secondary - bosster charge. Has optimal density interval, if density is too low burns - without detonation, if too high - becomes overpressed.
Used in safe electric detonators and futuristic light ignition detonator systems.
Synthesis of BNCP
1. Preparation of [Co(NH3)4CO3]2SO4*3H2O complex. 47g CoSO4*7H2O is dissolved it 125 ml of water and is added to
solution of 100g (NH4)2CO3 in 500 ml H2O + 250 ml 25% ammonia solution. Resulted dark violet solution is oxidised by addition of 14 ml of 30% hydrogen
peroxide. Solution is allowed to sit for 30 minutes, after that period it is placed on boiling water bath and is evaporated to 300 ml volume. During
course of the concentration process, solid ammonium carbonate (25 g) was added in installments. Solution is filtered from unsignificant ammount of
precipitated black cobalt oxide and is further concentrated to 200 ml volume. Solution is slowly cooled to room temperature, product precipitates as
small deep-red prisms. They are filtered out from the solution and allowed to dry at room temperature. Yield is 16 g of pure product, mother liquer
can be further concentated with addition of ammonium carbonate to get more 16 g of complex, but it is less pure.
2. Preparation of [Co(NH3)4CO3](ClO4) complex. 16g of complexed synthesised on first stege is dissolved in 320 ml of
water. Solution is filtered and 16g of sodium perchlorate in 40 ml of water is added with stirring. Mixture is cooled on ice (or in freezer) for 3-4
hours. Product precipitates as small, lustrous, sharp violet prisms. Crystalls are filtered off, washed with small ammount of ice cold water and dried
at room temperature. Yield is about 14 gramms.
3. Preparation of BNCP. 14g [Co(NH3)4CO3](ClO4) is dissolved in 140 ml 10% perchloric acid, and solution of 26.5g of
sodium 5-nitrotetrazolate dihydrate in 450 ml of water is added with stirring. Solution is placed on boiling water bath, and allowed to sit there for
4 hours. Solution is slowly cooled to room temperature and then cooled in freezer to 10°С, precipitated crystalls of BNCP are filtered of,
washed with cold water, and recrystallised from 1% perchloric acid. Yield is 12.9g.
Photos of reaction products: first is tetraminocarbonato cobalt (III) sulphate trihydrate (product 1), second is tetraminocarbonato cobalt (III)
perchlorate (product 2), third is crystalls of BNCP.
This is the video of deflagration-detonation transition of BNCP in paper tube. Sorry i know i have to attach this file to post, but it seems to bee
too large, i tried 2 times with no result. So i have to place it on rapidshare:
DDT of BNCP video
[Edited on 11-3-2008 by Engager]
chemoleo - 11-3-2008 at 17:12
Hello Engager, very very nice! Inorganic syntheses of beautiful compounds, yet with such interesting properties!
Recrystallised from 1% HClO4, is the compound unstable or why is this necessary? Did you base your synth on a published report (if so, pls say) or
work it out yourself? I imagine the former as you also cite a lot of det data.
I was wondering whether you would upload the video to utube, it's likely to last longer there, and at least I can download it there!
Engager - 12-3-2008 at 01:40
Recrystallization is for purification, it is not necesary but can be done if needed. And i forgot to post reaction schematic:
References are on Russian language, i attached them to the bottom of this post.
[Edited on 12-3-2008 by Engager]
Attachment: BNCP Russian References.rar (188kB)
This file has been downloaded 1643 times
Rosco Bodine - 12-3-2008 at 07:46
I would have guessed a different outcome for that last reaction which would essentially be a mixed salt of the
cation tetrammine Co dihydroxide complex , with one nitrotetrazolate anion and one ClO4 anion .
The same reactions should probably occur for copper and probably also for nickel .
I wonder , and have some skepticism actually , about
the need for the "carbonato transition" shown . It seems
the entire carbonate intermediate is unnecessary .
The reaction scheme seems so closely parallel to the reaction where tetrammine copper (di)perchlorate is precipitated from the double decomposition
reaction of concentrated copper nitrate solution added to a warm NH4OH solution of NH4ClO4 .
What I see as more likely for Cobalt compound III ,
is that central two hydrogens on the extreme right ,
which are associated with the hydroxyls , are *not*
supposed to be there at all .....but simply the two
hydroxyls which define the dibasic structure of the
tetrammine cobalt "dihydroxide" base . It is a base ,
*not* a dihydrate as shown .
The subcase "3" for the ClO4 group in the last line
should be 2 not 3 as shown .
And the final molecule should have the ClO4 group substituted for the lower nitrotetrazolate inside the
bracketed larger structure , with a subcase of *2*
outside the bracket .
That's the way I see it as more plausible anyway .
And one last thing , it doesn't seem possible that this
substituted tetrammine cobalt perchlorate would be stable in neutral or acidic aqueous solution , but only in an ammoniacal solution of mixed ammonium
nitrotetrazolate
and ammonium perchlorate . In aqueous solutions otherwise it should decompose via hydrolysis , the
same as does tetrammine copper perchlorate .
Being structured and reportedly behaving so differently
from what would be expected , it is certainly anomalous
in many ways if all of this is not true .
[Edited on 12-3-2008 by Rosco Bodine]
Engager - 12-3-2008 at 08:00
Yea, this all is not my invention, read refs i provided. Cobalt with ammonia forms Co(NH3)6 and NH3 can not be substituted with 5-nitrotetrazole,
carbonate is only a "protective" group, whitch is finaly destroed on final stage of synth to free 2 coordination places for NT. Tetramine complex is
stable in acid even in 30% HClO4 you can make it for yourself to see this is true, so all is correct, read more about cobalt complexes before making
your statements. Your are denying practicaly observed and documentaly patented way of synth, may be you just go on and got top Nobell chemistry award
if you are so clever.
[Edited on 12-3-2008 by Engager]
ooops !
Rosco Bodine - 12-3-2008 at 09:45
Pardon my brain fart above . I just went back and reread
the text description and equations and saw the use of H2O2 to change the valency of the Co +II to Co +III in the first reaction which then changes
everything to follow . The +III valency I was simply reading as "compound #3" and not paying attention it was the valency designation for the Co .
My Nobel prize will have to wait until I clean my glasses and have another cup of coffee
Interesting compound , and the reactions do make a lot more sense now .
Edit: I still have my reservations about the hydroxyls .
With a Co+III complex , should not we see a hexammine cobalt trihydroxide as the base ?
[Edited on 12-3-2008 by Rosco Bodine]
hokk - 19-3-2008 at 23:23
| Quote: |
Originally posted by Engager
Synthesis of BNCP
|
I don't know if these are stupid questions or not, but anyways:
Could I use CoCl2 instead of CoSO4 without any problem? (changing the added mass of course)
Could I also use (NH4)HCO3 instead of (NH4)2CO3 if I add more of the (NH4)HCO3?
Thanks!
[Edited on 20-3-2008 by hokk]
Engager - 20-3-2008 at 03:10
No, you can't use CoCl2 because it will form complex with different properties and structure. Synth of Werner complexes is quite delicate and require
good replication of synth procedure. You can use NH4HCO3, but procedure have to be modified. This synth is taken from literature, but if you modify it
to use NH4HCO3 it can work but also may fail because a different solubilities/concentrations, so tryout is needed.
Rosco Bodine - 20-3-2008 at 12:41
Cobalt nitrate and ammonium carbonate might work okay .
As for substituting the bicarbonate , it seems like that should work okay if you add extra NH4OH
sufficient for conversion of the acid carbonate to the normal carbonate .
Hmmmm....according to this article attached , the chloride
does form the analogous carbonatotetrammine cobalt (III) chloride ,
as part of a series of analogous salts derived
from different cobalt salts used as the starting material .
The efficiency for conversion to the perchlorate would be solubility related .
But it doesn't appear structure would be any issue .
Engager is likely incorrect about the chloride not being workable .
[Edited on 20-3-2008 by Rosco Bodine]
Attachment: Carbonatotetrammine cobalt (III) nitrate.pdf (91kB)
This file has been downloaded 1880 times
carbonatotetrammine cobalt (III) chloride
Rosco Bodine - 20-3-2008 at 20:10
There is a preparation of the chloride analogue
described in the attached article where it is an
intermediate used in further synthesis .
Attachment: carbonatotetrammine cobalt (III) chloride intermediate.pdf (124kB)
This file has been downloaded 2148 times
Engager - 22-3-2008 at 21:21
Recently i've got interesting idea how bis-tetrazole can be made without use of dicyan as starting product. I found rection in Elderfield book of
heterocyclic chemistry, there was a reaction of HN3 with oximes, it yelded corresponding tetrazole, so i thought to use this scheme to generate
bis-tetrazole from glyoxime. Starting products will be glyoxal and hydroxylamine, they condense readily to form glyoxime, whitch is then reacted with
2 moles of HN3 to yield 5,5'-bistetrazole:
However i have not found this synth in literature, but i think it can work. Literature methods use Mn to precipitate insoluble Mn bis-tetrazole salt,
but case above this can not be made because one don't want to allow Mn azide to form. Excess of azide can be destroyed by using acidic nitrite
solution on boiling, this will destroy azide: HN3 + HNO3 = (boiling) => N2 + N2O + H2O
Also there is one problem - low solubility of glyoxime in cold water, this forces to preform reaction in hot solution - this will generate a lot of
fumes of volatile hydrogen azide, what is dangerous.
Somobody got any ideas about this? Will this synth work, and how to rise solubility of glyoxime in cold water?
[Edited on 23-3-2008 by Engager]
convergence of earlier posts
Rosco Bodine - 23-3-2008 at 07:02
| Quote: |
Originally posted by Engager
I've also made two other tetrazole-based energetic compounds. Diazoaminotetrazole and dihydrazinium 5,5'-azotetrazolate (mentioned as substance with
highest known positive heat of formation).
Synthesis of sodium bis-5,5'-diazoamintotetrazolate from aminoguanidine bicarbonate
Prepare mixture of 11.5 ml 70% nitric acid with 100 ml of water, add by portions with stirring 24.8g aminoguanidine bicarbonate. Stir mixture until
CO2 evolution stops and then add 20.4 ml 70% acetic acid. Mixture is stirred and slightly heated until all solid dissolves. The resulting clear
yellow solution is solution of aminoguanidine nitrate in 12-13% acetic acid. This solution is cooled in freezer to 3-4C, well mixed and placed on ice
bath. Slowly, with stirring, by small portions at time ice cold solution of 17.5g sodium nitrite in 75 ml of water is added. While addition,
temperature must be all times kept below 12C, perfectly in interval of 5-7C, process takes about 30-40 minutes. After diazotation is finished mixture
is removed from ice bath and left to stand at room temperature for 24 hours. Some time after removal from ice bath slow evolution of nitrogen begings,
and mixture can heat up to 25C, this is ok, so don't worry, and after about 12-16 hours of standing evolution of nitrogen stops and large amount of
diazoaminotetrazole precipitates. Solid is filtered off, washed with ice cold water slightly acidified with acetic acid and left to dry at room
temperature. Yield is about 50% of pure mono-sodium salt of diazoaminotetrazole. Photos of product shown below:
Reaction scheme:
[Edited on 11-9-2007 by Engager] |
On page 2 of this thread , in the second post there is a similar synthesis reported in US2064817
which uses a sodium acetate - acetic acid buffer for the diazotization .
I wonder if the buffer system has been tried and compared
with your described method , to see if the use of the buffer
can improve the yield above 50% . Also , even though
it has been specified that a non-mineral acid buffer system
be used , I wonder if boric acid - sodium borate has ever been tried and found to be unworkable .
Related to your interest in the metal ammonia complexes of these tetrazoles ,
which I have been following with interest here , also on page 2 of this thread was reported a
Copper Ammonium Salt of Diazoaminotetrazole , described
in US2004719 . On page three you show a sample of the
non-complexed parent compound in matchbox #3 which
you called CuDAT .
| Quote: |
Originally posted by Engager
I've attached photo of silver 5-nitrotetrazolate, among other tetrazole derivatives i've made so far. Upper-left to lower-right: 1. Copper-ammonium
complex 5-nitrotetrazolate (NH4)2[Cu(N4C-NO2)4(H2O)2] - green primary (NH4CuNT) ; 2. (N2H4)2(N4C-N=N-CN4) - dihydrazinium 5,5'-azotetrazolate (HZT) ;
3. Cu3(N4C-N=N-NH-CN4)2 - copper 5,5' diazoaminotetrazolate (CuDAT) ; 4. Cu(N4C-NO2)2*(HN4C-NO2) - acid copper salt of 5-nitrotetrazole (CuNT) ; 5.
Ag(N4C-NO2) - silver 5-nitrotetrazolate (AgNT) ; 6. Na3(N4C-N=N-NH-CN4) - sodium 5,5'-diazoaminotetrazolate (NaDAT) ; 7. Na2(N4C-N=N-CN4) - sodium
5,5'-azotetrazolate (NaAT) ; 8. HN4C-NH2*H2O - 5-aminotetrazole monohydrate (ATZ).
[img]http://www.sciencemadness.org/talk/viewthread.php?action=attachment&tid=8144&pid=106891[/img] |
Your picture of CuDAT is not clear enough to be sure , but looks to be a crystalline lump material ,
whereas the patent US2004719 describes the CuDAT as being an amorphous powder .
I am curious about your method of synthesis for the CuDAT , and also am curious if you tried the complexing
with ammonia as described in US2004719 . There is only
mention of the usefulness of the ammonia complex as a
sensitizer component in priming mixtures where it is used as a small percentage .
Is there more information concerning the possible usefulness of the complex alone ,
or the parent compound CuDAT as an initiator ,
described in the Russian literature ? Have you done any experiments with either
the CuDAT or its diammine ammonia complex to see if these
compounds have usefulness as "green primaries" ?
[Edited on 23-3-2008 by Rosco Bodine]
Engager - 23-3-2008 at 07:15
Stickers on boxes may not be correct subtance with sticker Cu(DATZ)2 on box is copper diazoaminotetrazole, that's why i listed correct names above
picture. Actualy Cu diazoaminotetrazole is amorphous, but when it dried it stucked to paper in some kind of lumps, in wet state it's olive green, but
dry it is almost black. It's quite sensitive to friction, it exploded then i tried to grind it in mortar. Explosion was quite loud, but i don't find
any usefull applications for it in literature, only mentions about it's existance. Surely it is highly sensitive and powerfull explosive, but i'm not
sure about it's initiative power. I have not tried ammonium complex, because i think it's too sensitive.
I attached photo of wet product below:
[Edited on 23-3-2008 by Engager]
.jpg - 58kB")
Rosco Bodine - 23-3-2008 at 07:44
I was thinking that possibly the CuDAT or its diammine complex might substitute for lead azide
in a binary composition with barium styphnate , as a lead free primary . This binary is supposed
to have a greatly reduced mechanical sensitivity , excellent stability , good initiating power
and is cheaply made .
See US3284255
http://www.sciencemadness.org/talk/viewthread.php?action=att...
Another possible binary would be a mixture of the CuDAT or its diammine with guanylazide picrate .
These same binary mixture schemes might also be useful with some of the other
complexes which have been described , as a means of improving the economics
and lowering the sensitivity for the combined mixture .
Another possibly useful guanylazide is the styphnate ,
but I have found no mention of this in the english language literature .
Is there any mention of guanylazide styphnate
in the Russian literature ?
[Edited on 23-3-2008 by Rosco Bodine]
Engager - 28-3-2008 at 08:57
I've tried synth of bis-tetrazole from previous post, with no success. I've used following procedure: 10.7g of sodium azide and 13g of ammonium
chloride are dissolved in 150 ml of water, then 8.8g of glyoxime was added. Mixture is heated on water bath, until glyoxime fully dissolved and then
refluxed for 6 hours. No collor change occured, no glyoxime precipitated after cooling, then 16.8g of soda is added to destroy ammonium salts, and
solution is boiled on water bath for more 2 hours. Solution color changed from pale yellow to deep red, no product precipitated after cooling.
Unreacted azide is destroyed by action of nitrous acid: 25 ml of sodium nitrite is dissloved in 100 ml of water, and is added to red reaction mixture,
then solution of 20 ml conc. sulphuric acid in 140 ml of water is added by small portions. Destruction of azide is accompanied by virgous evolution of
nitrogen oxides, and foaming, addition of sulphuric acid was continued until mixture show acidic pH, iodine-starch paper test shown blue singnal
(means excess of nitrite in reaction mixture). Reaction mixture was allowed to sit for a night, and after very large prismic crystalls are
precipitated - longest one 7 cm lenth with 0.5 cm width. Presence of bis-tetrazole was analyzed by addition of manganeese (II) sulphate (Mn salt of
bis-tetrazole reported to be insoluble in water), no precipitate was obtained from mother liquer and from solution of precipitated crystalls. So
structure of product obtained is unknown, but it is not bis-tetrazole, because no precipitate with MnSO4. Structure of red intermediate in solution
during synth pathway is aslo unknown.
Today i thought another way to make bis-tetrazole without use of dicyan. It's simmilar to synth of 5-aminotetrazole by Thiele method. Glyoxime must
react with hydrazine forming corresponding hydrazone, whitch is "dimeric" analogue of aminoguanidine, diasotation with HNO2 should result in "dimeric"
guanylazide, whitch should form bis-tetrazole in way simmilar for guanylazide=>5-aminotetrazole. Proposed reaction scheme:
Any ideas about this possible path? Sobody have any information about intermediate products and their properties? Well be glad to hear any oppinions
about this synth path. The_Davster! What do you think about it?
[Edited on 28-3-2008 by Engager]
| Pages:
1
2
3
..
7 |



























