


 I hold the loop with a gloved hand--yes, molten NaOH is nasty-- and work it around the
melt to get the Na inside. I then lift it out and slap it against the test tube containing motor oil (just because it was nearby). I ran the setup
this way for an hour, removing the little balls of Na and quickly returning the loop to the melt. I was usually fast enough to get it back before the
NaOH solidified. I kept the propane torch on low flame nearby so if I was too slow, I could re-melt and be making Na within a few seconds. this
technique seems to work acceptably well (at least for me) to make small amounts of Na. I'm still amazed that a mere 35 watts in ( I'm
running this setup at 10V and 3.5 A) is enough to keep the melt at 315C without any attempt to insulate from ambient. I have not yet perfected a way to clean up the Na that is produced. I've
melted it under paraffin, xylene and ordinary paint thinner, all three of which result in shiny balls of Na but there is still come contamination
(NaOH) visible each time. Ideas welcome.
I hold the loop with a gloved hand--yes, molten NaOH is nasty-- and work it around the
melt to get the Na inside. I then lift it out and slap it against the test tube containing motor oil (just because it was nearby). I ran the setup
this way for an hour, removing the little balls of Na and quickly returning the loop to the melt. I was usually fast enough to get it back before the
NaOH solidified. I kept the propane torch on low flame nearby so if I was too slow, I could re-melt and be making Na within a few seconds. this
technique seems to work acceptably well (at least for me) to make small amounts of Na. I'm still amazed that a mere 35 watts in ( I'm
running this setup at 10V and 3.5 A) is enough to keep the melt at 315C without any attempt to insulate from ambient. I have not yet perfected a way to clean up the Na that is produced. I've
melted it under paraffin, xylene and ordinary paint thinner, all three of which result in shiny balls of Na but there is still come contamination
(NaOH) visible each time. Ideas welcome.| Quote: |
 I had to wash my mouth out and now I have a little burn. I used a diffrent PSU(5v
20a) and I saw a little sodium but I could not collect it. I think its redisolving in the NaOH even though I turned off the plate and lifted the spoon
off it when I applied the current.
I had to wash my mouth out and now I have a little burn. I used a diffrent PSU(5v
20a) and I saw a little sodium but I could not collect it. I think its redisolving in the NaOH even though I turned off the plate and lifted the spoon
off it when I applied the current.  The sad thing was that I had a multimeter
sitting on a bench 5 feet from me the whole time. I didn't get much sleep last night.
The sad thing was that I had a multimeter
sitting on a bench 5 feet from me the whole time. I didn't get much sleep last night.| Quote: |
| Quote: |
(see sodium acetate etc)


| Quote: |
| Quote: |
| Quote: |

| Quote: |
| Quote: |
| Quote: |
| Quote: |
| Quote: |
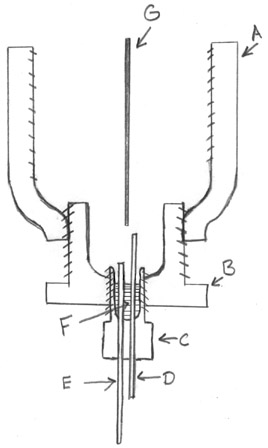
Now I know this isn't ideal and that it's probably going to
shortcut the cell when the molten sodium collects in it, but at the moment I have nothing better at my disposal. 


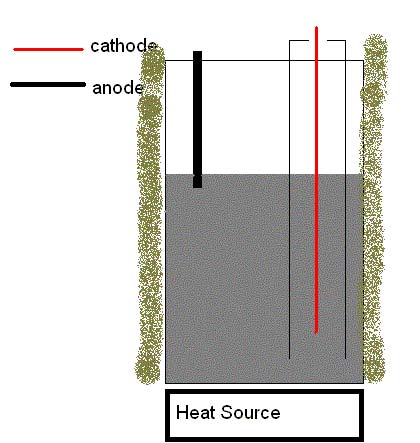
 There are
some transparent spots developing in the NaOH too....
There are
some transparent spots developing in the NaOH too.... | Quote: |
| Quote: |
| Quote: |
Now that's confusing, especially because of the thermite reactions that occur with lye and aluminum. (Tacho was
doing experiments with these)