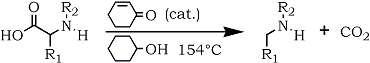@Ephoton,Yes Hofmann Rearrangement is really interesting

I think we can customize below route for Ethylamine .
This method for making ethylamine is easier than methylamine because ethylamine boil at ~16c( I have access to calcium propionate  ) )
But i think ammonium propionate can easily be made from ammonium sulfate and calcium propionate.but i dont know what temp needed for making ammonium
propionamide from ammonium propionate.
| Quote: |
Synthesis of Methylamine/Methylamine HCl via Hofmann Rearrangement
There are two approaches to producing an amine from an amide using the Hofmann rearrangement reaction. One way is to react the primary amide with an
alkaline-halide solution (eg - Sodium Hydroxide and Bromine). The other method is to use an alkaline-hypohalite solution (eg - Sodium Hydroxide and
Calcium Hypochlorite). The astute observer will notice that there is no chemical difference in the two processes. One produces the Hypohalite in situ,
the other uses the Hypohalite itself. Substitution of various halogens/halides/hypohalites/hydroxides is acceptable, but I feel I have picked the best
combination of maximal yield and ease of availability. Feel free to prove me wrong ;-). Also, at least one text specifies that Sodium Hypochlorite
produces much higher yields than Sodium Hypobromite. This delicious vindication is expected since Chlorine is a more reactive halogen than Bromine.
Now, we are going to use Calcium Hypochlorite, in the form of powdered pool shock, because concentrated Sodium Hypochlorite is a rare, unstable
creature indeed. Our yield will not suffer in the slightest because of this.
The main example presented will be the alkaline-hypohalite method as it is the easiest to acquire the necessary chemicals. It is of interest to note
that the alkaline-halide method is much easier to perform, processwise, in that it is more forgiving of sloppy technique.
The general theory behind the process is that the hypohalite will convert the amide to a haloamide. This then spontaneously changes to the isocyanate
when heated and decomposes to the amine from the water present. In effect, all that happens is that a Carbonyl (CO) group is stripped off the starting
amide to yield the corresponding amine. Yields pre-purification are around 80%, post-purification average around 65%. Certain uses of the resulting
amine will not require purification, though, so it will be left up to you whether or not to perform those steps.
To make methylamine we start with Acetamide. The general, unbalanced reaction process is thus:
CH3CONH2 + Ca(OCl)2 ----> (CH3CONCl)2Ca++ + H2O
then
(CH3CONCl)2Ca++ + NaOH ----> CH3NH2 + Na2CO3
CAUTION Methylamine is a poisonous, noxious inflammable gas. It has a strong ammonia/rotting fish-like odor. It's not as bad as Chlorine gas,
though, which can be produced if one is careless in the beginning!
You can scale these reactions up or down within reason. What is reasonable? I can't say, but I have done batches from .01 to 1 mole with no
difficulty. The key problems in scaling this reaction have to do with heat gradients in the flask and inadequate stirring. Use your own judgement,
keeping in mind that this is not an industrial process.
One reference to keep in mind (Thanks to J.W. Smith for sending this one) concerns the first step of the reaction.
Whitmore and Thorpe, J. of the Amer. Chemical Society, Vol 63, April 1941, p1118
"It was necessary to allow several hours for the formation of the N-chloroamide before heating to degradation temperature. With this modification it
was possible to prepare methylamine...consistently in 78% yield."
In my experience, this is a true statement. Please remember to keep the reactants well iced, though. Now, to begin:
In a large mixing bowl which can contain a smaller stainless steel mixing bowl, prepare an ice bath with water and salt to bring the temperature down
to -10C or so. Setup your glassware for simple distillation with magnetic stirring beforehand because certain steps need to be performed quickly. Use
a vacuum adapter to connect to the receiver flask, and attach some rubber or polypropylene tubing to the vaccum nipple to connect to a bubbler setup
(a funnel inverted in a beaker, or a plastic aquarium aerator tube). The distilling flask should be sitting in in a stainless steel bowl with nothing
in it (you will add pre-heated oil to the bowl).
NOTE In order to make this as painless as possible, please observe the following recommendations: 1) Keep the mixing bowl temperature as close to 0C
or less as possible; 2) Keep the Hypochlorite solution as it is being added as close to 0C or less as possible; 3) After half the Hypochlorite
solution has been added, place a plastic bag with 50-100g ice/salt/water mix into the bowl to help keep temperatures low (use this instead of directly
adding ice to the reactants, which adds a considerable volume of water making the process less volumetrically efficient); 4) Purchase an 8lb bag of
ice ahead of time!
Next you will prepare three solutions.
10g of Acetamide in 20mL of distilled water.
16.4g of Calcium Hypochlorite (Pool shock) in 50mL of hot distilled water
24g of Sodium Hydroxide (Lye) in 40mL of cold distilled water
This last solution should be prepared slowly as it is quite exothermic. Set all three aside in a freezer. Now prepare the mixing apparatus which will
be a stainless steel "mixing bowl" suspended in the ice/salt bath made earlier. We use a stainless steel bowl here so that heat transfer will be
maximal, while preventing any corrosive interaction. A glass bowl will not be sufficient for larger scale preparations as it will not conduct heat
fast enough to prevent the reactants from going over 10C (at which point the Haloamide will decompose and you'll have to start over). Take the Sodium
Hydroxide solution out of the freezer once it is cool, but not cold.
After the bowl has been sitting in the ice bath for a few minutes, add the Acetamide solution. Stir well until the solution has cooled to -10C. Now,
slowly add the Hypochlorite solution to the mixing bowl in bursts of no more than a couple mL while stirring vigorously. If you do this perfectly,
there will be no fizzing or bubbling at all. This depends on how cold you keep the mixture, and how slowly you add the pool shock! Realistically, the
considerable heat evolution of the reaction will make adding the last few mL a trying task! Keep an additional 50g of ice on hand to throw directly
into the mixture if necessary. This solution may evolve Chlorine gas so you should obviously perform this step under a fume hood or outside). Keep
stirring until it has calmed down and turned a turbid colorless to light green Let it sit for 2 hours, stirring occasionally and making sure that it
never gets warmer than 5C.
After the 2 hours is up, add the Sodium Hydroxide solution quickly with stirring. The solution should immediately turn a chalky, milk white. That's
because a lot of Sodium Carbonate just got generated. You no longer need be concerned over it's temperature, so you can leave the solution in this
state overnight if perhaps the hours have passed by too quickly and you've suddenly realized it's 2:00am.
Preheat a water bath on the stove (or wherever) to about 80C and place the stainless steel mixing bowl in it. Once the temperature of the solution
hits about 65C, take the bowl out and set aside while stirring all the while. This is where it rearranges, and the reaction is exothermic enough to
sustain it's temperature nicely. If you find the temperature climbing past 80C, immerse the bowl into some cold water briefly. After about 15 minutes
the temperature will start to fall, at which point you should transfer the whole mess to the distilling flask. Before you continue you need to choose
whether you want to make the hydrochloride salt or the aqueous solution of Methylamine, though.
Heat the flask using an oil bath to 100C after adding this solution to effect gentle boiling which will drive off the Methylamine as a gas. In my
experience, misbehavior is likely to occur at this point. One particular problem to watch out for is the sucking back of bubbler solution (be it plain
water or 6N HCl) into the receiver flask. I don't know why the pressure in the distilling flask would go below atmospheric, and therefore cause this
to happen, but it has several times with me. Needless to say, this results in a serious mess and botches the whole process (I have found a cure for
this by using an automotive one-way vacuum valve, like a PCV).
Continue heating the flask contents until you have collected around 100mL of distillate in the receiver.
For the aqueous solution: Place 18mL of cool distilled water into your bubbler setup. The expected, not theoretical, yield of Methylamine from this
amount of reactants is 7 grams. I have used a plastic aquarium aerator tube as the bubbler with excellent results. Sure beats using an inverted
funnel.
For the HCl salt: Do exactly as above except use 6N Hydrochloric Acid. 6N HCl may be produced by diluting 60.4mL of "Muriatic Acid" to 100mL with
distilled water. Evaporate the bubbler solution to dryness then add 15ml of water, 10mL 10% NaOH soln. and heat gently to a boil with constant motion
until dense white fumes appear. This will remove the Ammonium Chloride. Remove from heat while stirring as it cools down. Pulverize the dry residue,
then reflux with absolute Ethanol for several minutes. Filter the refluxed soln. on a heated Buchner or Hirsch funnel, then distill the alcohol off
the filtrate until crystals just begin to form. Allow the soln. to cool naturally to room temperature, then cool further in an ice bath. Filter the
solution on a chilled Buchner funnel with suction. The yield of Methylamine Hydrochloride should be around 55% of the theoretical.
To clean the white residue off of your glassware, dump some muriatic acid straight from the jug onto them and swirl.
References:
Journal of Chemical Education, v14, pg542
Organic Reactions volume 3
Vogels Elementary Practical Organic Chemistry, pg574
|
Do you know decompose temp of chloride salt of Ethylamine ?and solubility in acetone and EtOH?
[Edited on 21-1-2012 by Waffles SS] |







 Ain't no issues with
the synthesis of that shit...
Ain't no issues with
the synthesis of that shit...