
night429 - 8-5-2021 at 15:48
So, I've decided to try my hand at making an azo dye between these two compounds, just because I never seen it photographed at all. It also serves as
a sort of project introduction, as I'm planning to characterize additional ""obscure"" azo dyes in the future (I'm going to have multiple dyes in one
or two posts as to not spam topics, though). Now, into the introduction of the synthesis:
Because of the extremely low efficiency of the SNAr reaction of 2,5-dichloronitrobenzene with NaOH, I only had less than 100mg of the
4-chloro-2-nitrophenol (in the form of the sodium salt), and this was the smallest scale I've ever worked on.
To start, I weighed out 25mg of the red-colored sodium 4-chloro-2-nitrophenoxide. In a separate container, I weighed 330mg of sodium hydroxide and
added it to 3.5mL of water. After it dissolved, I added the solution to the sodium 4-chloro-2-nitrophenoxide and used a pipette to try and dissolve
it. I ended up having to add more water (6.5mL) to get it all to dissolve. At the end, the solution was a yellow-orange. I poured it into a flask and
set this aside.
After this, I weighed out 160mg of p-aminophenol (obtained from the acid hydrolysis of acetaminophen). With stirring, I added this to 5mL of water. I
dripped in 31% HCl until the solution cleared (quite a bit of bubbling occured, which I believe to be from a small amount of sodium bicarbonate
impurity). At this point, the solution was a green-ish. I put this in an ice bath with stirring.
I then weighed out 100mg of sodium nitrite and 1mL of water to it. Once it all dissolved, I waited until the p-aminophenol solution was cooled. It
leveled out at about 7°C, so I started adding sodium nitrite slowly, keeping the temperature below 10°C. The solution color dulled and turned (what
I would consider to be) "grayish-green".
I then took this out of the ice bath and took out the stir bar. I put the sodium 4-chloro-2-nitrophenoxide flask into the ice bath and then put in the
previous stir bar (after rinsing it). Once the temperature got to 7°C, I started to add the formed 'p-hydroxybenzenediazonium chloride' solution at
about a drop a second, keeping the temperature below 10°C. The solution took on a deep red color and had a rubbery smell (which I believe to be small
amounts of p-benzoquinone formed from the substitution of the diazonium group in the basic solution to hydroquinone, followed by oxidation by the
air).
After several minutes of stirring in the ice bath, I took it out to warm it up to room temperature while continuing to stir. The color started to turn
a bit brown, and I got slightly concerned and corrected for pH (I knew it would be very basic, but I was concerned that it was degrading the product.
Plus, I knew the phenoxide groups would increase solubility). So, I added 31% HCl dropwise until the pH was very acidic. At this point, I noticed a
precipitate. So, I filtered it off, soaked up excess water with a paper towel underneath, then dried it fully with a fan.
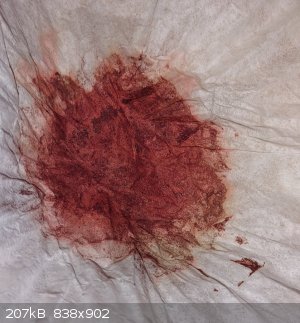
The yield I measured was 11mg, which represents a yield of 26%. While this isn't great, the only reason I made this compound was to characterize its
optical properties, and there are quite a few things I was pretty lax on (such as getting the temperature fully down to around 0°C, sort of
improvising the NaOH amount, etc.).
The end product bears a resemblance to matchbox red phosphorus. It's nearly black without strong light.

But, in the sunlight, you can see the purple-red color of the product (you might need to zoom in, but it's there to at least some degree).

Also, the color was very prominent on the filter paper remnants (most likely because the product that was trapped on/in the filter paper was already
very fine.
Fery - 8-5-2021 at 23:03
Hi night429, well done !!! Keep up your good work!
The temp 0 C is achieved by addition of ice directly into the diazotisation reaction mixture, low temp prevents diazonium salt decomposition. Above 5
C diazonium salts may decompose (sometimes even violently or explosively), releasing N2 and corresponding phenol is formed. Below 0 C there could be
some undesirable crystallization of diazonium salts. But for some reactions, even temp. above 5 C did not cause problems, chemists at Lundbeck performed such thermal studies, it was Sandmeyer reaction, no issues found at T range 5-15 C.
So you obtained p-aminophenol from acetaminophen pills, easiest way and enough for this scale. For larger scale, nitration of phenol is quite messy (I
tried it, only nitration part, not yet the following reduction, especially purification of the p-nitrophenol was tricky and required too much work),
maybe cleaner way is phenol->p-nitrosophenol->p-nitrophenol (I did not try it)->reduction, or nitrobenzene + Zn + aq. NH4Cl
->phenylhydroxylamine->p-aminophenol (Bamberger rearrangement)
[Edited on 9-5-2021 by Fery]
night429 - 9-5-2021 at 08:13
Hey Fery,
Thank you! I was unaware that temperature could be quite higher than the temperatures typically used. I did use p-aminophenol from acetamenophen for
this reaction, and I used a one-pot reaction starting from the Tylenol pills directly. I had an accident and spilled the majority of it, so I couldn't
determine a yield of this. I later redid it, and got a yield of 278%!! So, my p-aminophenol is extremely dirty (with the biggest impurity being sodium
bicarbonate, I believe), and I'm going to try and purify it. I tried a water recrystallization, but because I didn't squeeze the water out after
filtering, I had significant oxidation, so I'll see what I can do.
The procedures you had listed seem pretty interesting to do in order to get pretty pure p-aminophenol. I've been avoiding making phenol because I
don't have a very good way to get the temperature to the 200°C+, and high-temperature reactions scare me a bit as well. But, I should at least try it
(especially because I need o-nitrophenol for a separate reaction).
Fery - 9-5-2021 at 10:53
Hi night429, pills are good source of very pure p-aminophenol. Maybe you could try to extract p-aminophenol from pills using an organic solvent in
which the additional substances won't dissolve?
For big scale I do not suggest phenol nitration route, I did it once and the o-nitrophenol obtained was quite trivial and easy, the p-nitrophenol
quite tricky. The nitration of phenol was the most messy reaction I have ever done, it was quite surprise for me that I obtained the 2 isomers.
I definitely suggest the nitrosophenol route instead which gives only para isomer which could be oxidated to p-nitrophenol (but this oxidation is
contraproductive and unnecessary) and then reduced to aminophenol, or the nitrobenzene route to phenylhydroxylamine and rearrangement.
Anyway I have some p-nitrophenol put aside (last crop of crystals which is not so pure as main crop of crystals) which I plane to reduce to
p-aminophenol when I have time and all higher priority syntheses are done.
[Edited on 9-5-2021 by Fery]
night429 - 9-5-2021 at 12:45
Hey Fery,
Okay, I'm glad to know the purity can be quite high from pills. I may try to redo the recrystallization from water or dissolve the p-aminophenol in
acetone to leave behind any salts. The product that I have is decently free from organic contamination, since the pills I was using are around 90%
acetaminophen, and the method of extracting the p-aminophenol is decently selective (soluble at low pH, insoluble at pH of around 8).
I'm looking forward to reading about your p-aminophenol reduction when you get around to it!
Fery - 14-5-2021 at 05:14
here I found the complete synthesis of p-aminophenol from nitrobenzene:
https://labmonk.com/synthesis-of-p-aminophenol-from-nitroben...
pumpkins33d - 15-5-2021 at 21:21
Hey night429,
what about extracting the acetaminophen with acetone and then boiling off the solvent if nothing else is soluble in acetone that is. I looked at some
pill brands and the ones I found have some other ingredients that maybe soluble in acetone and water so a quick water washing may remove them. Or a
recrystallization of the acetaminophen from water once it has been extracted.
night429 - 16-5-2021 at 17:45
Hi pumpkins, I should really try that. I thought that the one-pot would have been less work, but it gives a very impure product (I got nearly a 300%
yield once). I'll redo it using 91% isopropanol, which I have more of, and with the pure acetaminophen, the p-aminophenol should be a lot purer than
not.
I did have success recrystallizing the impure p-aminophenol with water and a very small amount of sodium bisulfite as an oxygen scavenger (it needs a
hot filter before cooling). Once cooled and filtered, the water needs to be squeezed out as fast as possible (I used paper towels), but once pretty
much all of it is gone, it doesn't oxidize nearly as fast and can be dried with a fan. I haven't tested its properties as of yet, but considering it
hasn't gone a dark purple, it hasn't oxidized too much.
Here's my recrystallized p-aminophenol (still very slightly damp):

Also, thanks for the procedure Fery!
Boffis - 16-5-2021 at 20:02
Hi night429; I have found that it is better to store p-aminophenol as its hydrochloride. It can be obtained directly as fairly large crystals that
oxidized much less quickly. Its not stable indefinitely but it is easily handled over a period of weeks rather than seconds before is discolours
further.
pumpkins33d - 16-5-2021 at 20:08
I went to the store and tried a solvent extraction from some acetaminophen pills that had the least amount of inactive ingredients. I got a good yield
and the crystals looked very pure although I didn’t do any tests to verify purity. I got an 85% yield from just boiling the crushed pills in acetone
then hot filtering and boiling off the solvent.

night429 - 16-5-2021 at 20:51
Huh, I've found that p-aminophenol doesn't discolor at all when stored as the freebase... just stays brown once it's dry, even when stored for a while
(doesn't turn purple or black). Perhaps the oxidation doesn't cause a color change, though, and it's trash now. I'll see if I can make the
hydrochloride via bubbling HCl gas through the p-aminophenol in heptane, though. I'm planning on using it for the infamous Skraup quinoline synthesis,
so maybe the sulfate would be better for that (I assume I could just drip it into an acetone solution of p-aminophenol). I'll report my findings for
both once I do it.
Those crystals look great pumpkins, nice and fluffy! I'm doing a "two-pot" synthesis of p-aminophenol right now. In the future, I'll do a similar
synthesis to you and just stop at actetaminophen, since it's nice to have on-hand.
Boffis - 16-5-2021 at 22:44
Hi night429, have you looked at this thread?
http://www.sciencemadness.org/talk/viewthread.php?tid=27849#...
See my entry of the 30th Jan 2014
night429 - 16-5-2021 at 23:44
Thanks Boffis, I'll have to try that instead for the hydrochloride! I haven't done too much research into this area, as you can probably tell.
Fery - 17-5-2021 at 04:14
Hi night429, nice idea of Skraup quinoline synthesis. So from p-aminophenol do you obtain 6-hydroxyquinoline? I'll plane
aniline+nitrobenzene+glycerine=quinoline, 2-aminophenol+2-nitrophenol+glycerine=8-hydroxyquinoline.
quinoline+KMnO4 (analogously as naphtalene oxidation) = quinolinic acid (pyridine-2,3-dicarboxylic acid)
night429 - 17-5-2021 at 08:49
Hey Fery, Yep, you do get 6-hydroxyquinoline! I'm following the 8-hydroxyquinoline prep from Vogel, but I'm gonna try to use nitrotoluenes instead of
o-nitrophenol because they're easier to make (I thought about using p-benzoquinone, but it would condense in sulfuric acid).
Those sound like good procedures! Do you have any further plans for the quinolines and quinolinic acid?
Fery - 17-5-2021 at 12:24
Hi night429 I will offer quinoline and 8-hydroxyquinoline to my friend Bedlasky, he can prepare various interesting complexes, he is much more skilled
in this than me. I plane to perform quinoline + KMnO4 -> quinolinic acid and perform some experiments with it... whether it is zwitterion so the
-COOH in position orto binds to N in pyridine ring so then the acid cannot make intramolecular anhydride like phthalic anhydride. Then maybe it can
create some interesting complexes too?
according these links:
https://www.sigmaaldrich.com/catalog/product/Aldrich/P64405?...
https://www.chemicalbook.com/ChemicalProductProperty_EN_CB52...
it creates intramolecular anhydride like phthalic anhydride,
according this link:
https://echa.europa.eu/substance-information/-/substanceinfo...
intermolecular (2 molecules bound together, but suprisingly with -COOH in orto position, not the meta position as I theoretically expected as the orto
-COOH could create inner salt with basic N in pyridine ring).
Btw the separation of orto nitrophenol is not difficult, steam distillation (lazy method without external source of steam, just boiling water +
mixture of nitrophenols in one flask):
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
night429 - 17-5-2021 at 13:03
It's interesting that there's the possibility of both inter- and intramolecular anhydride formations, and I'm curious to see what affects that and if
one is more favored over the other.
As for the o-nitrophenol, I was moreso saying nitrating toluene is a lot easier and higher yielding than having to make phenol from aspirin and
nitrating that. I already cooked a stir bar when I attempted the decarboxylation, which only ended up with less than a gram of phenol (of a
theoretical 6.8).