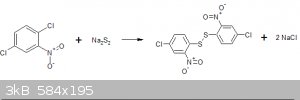
Preparation of Di-(4-chloro-2-nitrophenyl)-disulphide
An intermediate for the preparation of 4-chloro-orthanilic acid
Boffis November 2020
Introduction
This procedure was developed from the well-known synthesis of orthanilic acid (2-aminobenzene sulphonic acid) from 2-nitrochlorobenzene 1). This acid
cannot be prepared by direct sulphonation of aniline as sulphanilic acid, the 4-isomer, can. While the 4-chloro-orthanilic acid may be possible by
direct sulphonation of 3-chloroaniline no such procedure has been found and it is likely that the product would be a mixture of isomers. Orthanilic
and 4-chloro-orthanilic acids are used to prepare azo dyes such as sulphonazo III, a reagent for the detection of barium. The preparation of
4-chloro-orthanilic acid provides a useful derivative from p-dichlorobenzene via its mono-nitro-derivative.
The reaction involves the replacement of an "activated" chlorine group ortho to the nitro-group with a disulphide ion generated insitu from sodium
sulphide and powdered sulphur.
Experimental procedure:
70.01g of moist sodium sulphide nona-hydrate were stirred into 250ml of ethylene glycol. When most of the sulphide salt had dissolved and only a
little white solid remained 9.00g of powdered sulphur were added.

Fig. 1 Left; Sodium sulphide crystals and Right: Sodium sulphide dissolving in glycol
The mixture was heated and magnetically stirred until the sulphur had dissolved to give an orange brown disulphide solution.

Fig. 2 Left; sulphur dissolving in the glycol solution forming orange brown disulphide ions and Right; the final disulphide solution
To this were added fairly quickly a solution of 100.00g of 2,5-dichloronitrobenzene dissolved in 200ml of industrial methylated spirit with vigorous
stirring.

Fig. 3 Left; the 2,5-dichloronitrobenzene and methylated spirit and the nitro compound solution
The solution immediately turned almost black and then dark brown. After a few minutes a pale ppt began to form. The solution was stirred and heated on
a hotplate, set at 120°C, for 2½ hours and then the heat was turned off but stirring continued until the suspension had cooled to room temperature.
The suspension slowly faded from a dark brown to a deep orange yellow slurry.

Fig. 4 Left; the sulphide and nitro-compound solutions on mixing T=0 and the reaction mixture after 15-20 minutes, the product starting to precipitate
The cooled suspension was filtered at the pump, 50ml of industrial methylated spirit being used to rinse the flask and then wash the filtered cake.
The Buchner flask was then emptied and the filtrate kept to attempt to work up for further product. The cake was washed on the filter with two 50ml
portions of industrial methylated spirit and sucked dry as dry as possible. The cake was dried to constant weight at about 40°C, about 4 hours, to
give 67.07g of crude product.

Fig. 5 Left the final reaction mixture after 2½ hours and the final washed and dried product
The crude compound was stirred into 200ml of methylated spirit, warmed slowly to 45-50° with stirring and then cooled to room temperature .The bright
yellow solid was filtered off and washed with 2 x 50ml portions of methylated spirit after which the filtrate was practically colourless . The cake
was sucked as dry as possible and then dried at about 40° to give 62.57g of bis(4-chloro-2-nitrophenyl)disulphide or about 63% of theory. The
compound is pure enough for the preparation of the sulphonyl chloride.
The compound may be recrystallized from acetone or butanone, about 8ml per gram of the latter are required, to give a bright yellow crystalline
powder.
Attempts to recover more product from the initial filtrate resulted in only a brown oil from which nothing much could be isolated, certainly not
further disulphide.
Discussion
I have carried out this preparation several times and it is very reliable providing the sodium sulphide is of good quality glassy, colourless crystals
of the 9 hydrate. The yield and quality suffer if sodium sulphide flakes are used; the flakes are not anhydrous but contain about 3 to 3.5 H2O and
dissolve slowly in glycol although the addition of an appropriate amount of water helps. If the initial glycol solution is not clear the small amount
of white residue is sulphur and it is of no consequence.
The digestion of the crude product in hot alcohol is important as it removes any unreacted dichloronitro- compound.
I tried to prepare the same acid (4-chloro-2-aminobenzene sulphonic acid) from 2,5-dichloronitrobenzene by direct displacement of the 2-chloro group
with sodium sulphite. The reaction worked but the yield of the intermediate nitro-sulphonic acid was very poor, about 8% of theory. This procedure was
adapted from a procedure for the preparation of 2,4-dinitrobenzene sulphonic acid 2) from 2,4-dinitrochlorobenzene. I have run this procedure several
times, it works well and is high yielding but I could not selectively reduce the 2-nitro group to give 4-nitro-orthanilic acid.
Work is in hand to develop a procedure to convert the disulphide to the sulphonyl chloride without cylinder chlorine.
The disulphide link is easily reduced to a mercapto group and this is likely to occur if the nitro group is reduced giving rise to the possibility of
preparing other derivative from this intermediate such as 4-chloro-2-aminothiophenol.
1) Vogel; Textbook of Organic Chemistry; 5th edition
2) Fierz-David and Blangey; Fundamental Processes of Dye Chemistry; 1942; 5th Edition English trans.
[Edited on 24-11-2020 by Boffis]
