
UnintentionalChaos - 29-12-2008 at 21:12
So, ever since mellitic acid synthesis came up in this thread: http://www.sciencemadness.org/talk/viewthread.php?tid=10863&... I've been contemplating better ways to make it beside electrolysis or by the long
refluxing with nitric acid of various kinds of carbon. Another procedure outlined was the simple oxidation of hexamethylbenzene by permanganate, a
synthesis of which exists here http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv4... However, the method involves a heated catalyst tube, which is beyond what many of
us can probably set up.
To this end, I propose the following general procedure:
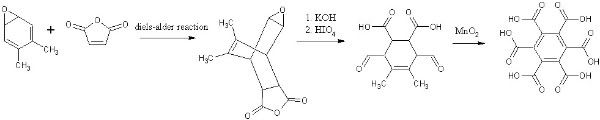
Of course, the final oxidative aromatization may require the use of sulfur or another similar reagent, but for now, I'll leave just the MnO2, which I
don't think will cleave double bonds. If it can, like permanganate, there are numerous other options. The specifics of the diels-alder would need to
be worked out, and most importantly, how to make the diene. What I'd like to know, is if there are any glaring problems that I'm not seeing, and any
thoughts about the diene of course. Lastly, do you think a vicinal diol would be a problem during the diels-alder? If not, then the epoxide cleavage
can be cut out (assuming the diol is easier to make), giving:
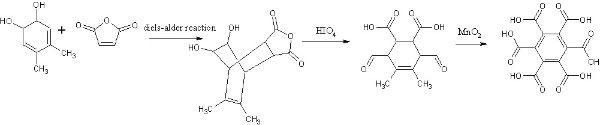
EDIT: The only reason for the epoxide instead of the diol is the possibility of making the diene like so:

[Edited on 12-30-08 by UnintentionalChaos]
sparkgap - 29-12-2008 at 22:18
Why do I have the sinking feeling that the final diene in the scheme in your edit will merely decompose to o-xylene? 
sparky (~_~)
UnintentionalChaos - 29-12-2008 at 22:44
Hrm. By extrusion of water, sparky?
Looking at it now, I'm gonna say the diol scheme is impossible. That'll probably dehydrate if you look at it too hard to 3,4-dimethylphenol.
[Edited on 12-30-08 by UnintentionalChaos]
sparkgap - 29-12-2008 at 22:50
I don't really know. Searching around using the IUPAC name of your epoxidized diene doesn't give me meaningful results, so I can't definitively say
that that epoxidized diene exists or otherwise.
However, my intuition is telling me that the bromohydrin in the second-to-the last step of your scheme in the edit will do anything it can to turn to
something with an aromatic ring. That would either be a xylene or a xylenol.
sparky (~_~)
UnintentionalChaos - 29-12-2008 at 23:44
Hrm. I was aiming for a locked s-cis conformation diene since that would be very reactive, but since the maleic anhydride is very reactive itself,
perhaps a non-locked diene would also be fine. Seeing as how I can't think of any way to make the epoxide anyway, how about this:

Any thoughts for ???. Id really rather not use a witting between 2-halobutane and methylethylketone, which would require PPh3 as well.
[Edited on 12-30-08 by UnintentionalChaos]
JohnWW - 30-12-2008 at 03:35
Those reagents and precursors are relatively expensive, compared to the known method of preparation of mellitic acid, also known as graphitic acid or
benzenehexacarboxylic acid, simply by the action of either alkaline KMnO4 or hot concentrated HNO3 on graphite, or of hot concentrated sulfuric acid
on charcoal.
The stuff was first discovered in 1799 by M. H. Klaproth in the mineral honeystone, or mellite, its aluminium salt, which is formed by the action of
water, air, and heat on coal deposits in the presence of clay minerals. Its anhydride C12O9 is in fact an oxide of carbon. See http://en.wikipedia.org/wiki/Graphitic_acid , http://dic.academic.ru/dic.nsf/cide/109414/Mellitic
Here is a patent for its industrial preparation:
http://www.freepatentsonline.com/2176348.html
It can be decarboxylated to pyromellitic acid:
http://www.orgsyn.org/orgsyn/pdfs/CV2P0551.pdf
Its being a polycarboxylic acid has led to examination of its use as a metal chelating agent, and in making hard polymers with multiple polyester
links.
Ebao-lu - 30-12-2008 at 06:10
This epoxide is the same as oxepin (they are in equilibrium), and you're quite right about its synthesis(elimination of HBr), but still i dont
understand how to make the first precursor. There should be some procedures in literature.. This epoxide should react rather well in D-A reaction ( at
least, unsubstituted oxepin reacts with maleic anhydride at room temp)
sparkgap - 30-12-2008 at 06:27
Ah, Ebao-lu, thank you for reminding us of the oxepin-epoxide equilibrium.  With
that in mind, I can no longer see how to proceed to mellitene from the Diels-Alder adduct between the oxepin and maleic anhydride.
With
that in mind, I can no longer see how to proceed to mellitene from the Diels-Alder adduct between the oxepin and maleic anhydride.
sparky (~_~)
Ebao-lu - 30-12-2008 at 07:34
I call it oxepin, because it is hard to find in literature by "epoxide diene", thats all. But in most reactions it still reacts as its epoxide diene
(D-A also). I mean, the desired product is formed at r.t. with maleic anhydride.
In order to make a non-locked diene, probably it is possible to start from methyl ethyl ketone and bind it to pinacone of formula
3,4-dimethylhexandiol-3,4 (with Mg), tosylate it and eliminate TsOH, or distill the pinacone over Al2O3, but in both cases i am not sure the desired
product would form.
Just was surprized to learn, that 48% HBr does not catalyse the pinacone-pinacoline rearrangement like H2SO4. It was written that heating of acetone
pinacone with 48% H2SO4 leads to diene. Hopefully, in case of methyl ethyl ketone the thermodinamical product would form in same reaction
[Edited on 30-12-2008 by Ebao-lu]
[Edited on 30-12-2008 by Ebao-lu]
UnintentionalChaos - 30-12-2008 at 10:59
Ebao-lu: thank you for reminding me of the pinacol coupling! It slipped my mind as I was looking at aldol condensations last night, which leave an
unwanted ketone group and would then require Br2 treatment after its removal. I'm sure something can be done to avoid the pinacol rearrangement. With
MEK as a feedstock, it'll be plenty cheap enough to tinker with. The McMurry reaction followed by Br2/dehydrohalogenation also seems viable (albeit
slightly more complex), if the rearrangement tuns out to be hard to avoid.
JohnWWW- yes, the reagents may be a bit more expensive, but if we proceed by the non-locked route, then it is well within the scope of members here.
Keep in mind that the oxidation of graphite isn't a clean sysnthesis. If you look at the numbers, 80 hour to one month reflux with nitric acid is
classically producing 1% or less yield. The procedure in the paper Axt posted in that last thread gives 10-11g from 100g of graphite, but after !!14
days!! reflux with a large excess of fuming nitric acid and 7 days in alkaline KMnO4. The products are refined by electrolysis. Who here feels safe
refluxing fuming nitric acid for 2 weeks straight? (Knowing the crowd, I'm sure somebody would). You need to consider time in making that fuming
nitric acid in the first place and if you really want to use it up for a low yield of C6(COOH)6. Overall, I think the traditional procedures are
sloppy and the slight increase in cost (if any) for reactants is well worth it, hence "logical" synthesis. There is a beautiful logical synthesis of
buckminsterfullerene out there, whose purpose is to avoid the long refining of soots to make pure C60, and this synthesis is in the same vein.
Mellitic acid esters
Boffis - 9-5-2011 at 06:33
Hi, this thread seems a little cold but I'll add my comments anyway!
I recently acquired a jar of sodium diethyl oxaloacetate along with various other chemicals. It sat for a while on one side because I couldn't think
of a use for it. However, I notice that the free ester would contain both an active methylene group and =O on the adjacent carbon atom which raises
the possibility of self condensation via a knoevenagel tpye condensation under the influence of an acid catalyst. The secret would be to get it to
trimerise rather than polymerize to a resin. The product would then be the hexaethyl ester of mellitic acid.
So when I get home I am going to try the following experiments:
1) simply dissolve in water and add sufficient HCl to the boil solution to neutralize the sodium salt, liberating the free ester with sufficient
excess HCl to catalyse the knoevenagel condensation. The hope would be that the ester groups remain intact and the hexa ester can be extracted with
ether or similar.
2) Just neutralise with HCl in the cold and extract with say CCl4 and pass dry HCl into the solution.
Either way the final mellitic acid would be liberated by alkali hydrolysis and then ppt with acid.
When I return home in mid June I'll give both a try and report.
Any one got any experience of condensations with this compound?
PHILOU Zrealone - 17-5-2011 at 04:21
Other ways would be:
Cyclisation (cyclopolycondensation) of R-O2C-CH2-CO-CO2-R (like Boffis proposed)
Cyclisation (cyclotrimerisation) of R-O2C-C#C-CO2-R
Cyclisation (cyclopolycondensation) of NC-CH2-CO-CO2-R and hydrolysis
[Edited on 17-5-2011 by PHILOU Zrealone]
PHILOU Zrealone - 19-5-2011 at 03:14
And last but not least another interesting synthesis of the stuff... from cyclopentanone. 
By trimerisation in basic or acidic media of cyclopentanone leads to a tetracyclo compound, an aromatic ring with 3 alcanic exo-rings (by propane
lateral chains) between position 1-2, 3-4 and 5-6 of the aromatic core.
3 (-CH2-CH2-)2C=O --> C6(=C3H6)3 + 3 H2O
Then by acido-permanganic oxydation (or bichromic oxydation) of all the lateral propane bridges...one ends up with mellitic acid.
C6(=C3H6)3 -Oxydation/H2O-> C6(-CO2H)6 + CO2 + H2O
[Edited on 19-5-2011 by PHILOU Zrealone]
Boffis - 19-5-2011 at 18:44
Has anybody tried preparing he precursor hexamethylbenzene by simply refluxing sulphuric acid with butanone (MEK) in a fashion analagous to the
preperation of mesitylene from acetone? This is again a knoevenagel type condensation and it might be possible to bring about the same reaction by
passing butanone vapour + nitrogen over alumina or porcelian at 300-350 C. This is basically the same reaction as Philou's above but with a more
available precursor.
Going back to Philou's earlier suggested routes the nitrile ester may be possible say via ethyl beta cyanopyruvate but how do you get this? Could it
be prepared in a fashion similar to my current possible precursor (ethyl acetate + Na ethoxide + diethyl oxalate = Na diethyl oxaloacetate) from say
acetonitrile, sodium ethoxide and diethyl oxalate? Might this polymerise the nitrile group?
I also have some natural mellite so I'll also try the extraction from this source too.
Looks like I'm going to be busy when I get home!
PHILOU Zrealone - 20-5-2011 at 02:44
I'm not sure the MEK synthesis way of mellitic acid would work fine...because the attack will more likely happen on the free CH3 side leading mostly
to 1.3.6-triéthylbenzene...aside maybe some hexamethylbenzene and other polymerisation products.
That is the reason why I had the idea to introduce an electronwithdrawing group like HO2C-, N#C-, R-O2C- to favourise attack of the carbonyle on that
specific carbon and also because there is no other to interact with... so in principe N#C-CH2-CO-CH3 must be another way to the desired product via
1.3.5-trimethyl-2.4.6-tricyanobenzene, oxydation and hydrolysis. Cyanoaceton (probably very lacrymating) is obtainable from chloraceton (also a
lacrymator).
Beware that the polymerisation via condensation of such stuffs might be very exothermic and even explosive if product are highly concentrated (the
heat has not the time to dissipate and increasing viscosity will reduce heat transfer...if heat goes too high cracking and fire may result...so better
keep the substances dilluted and fast used.
The obvious way to N#C-CH2-CO-CO2H is via lactic acid/pyruvic acid:
CH3-CHOH-CO2H -ox-> CH3-CO-CO2H
CH3-CO-CO2H -Cl2/H(+)-> Cl-CH2-CO-CO2H
Cl-CH2-CO-CO2H -CN(-)-> N#C-CH2-CO-CO2H
Note that pyruvic acid and intermediaries are very prompt to lose their external carboxyle into CO2 while moderate heat is set on (arround 100°C)
...so in all cases the system must remain cold enough but not too much in a way the reaction stil occurs...thank the help of the withdrawing group the
reaction can be done at low temp.
But there are 1000 others if you know chemistry of common molecules like ethanol, ethanal, glycol, glycerol, ...
[Edited on 20-5-2011 by PHILOU Zrealone]
Boffis - 13-7-2011 at 11:42
Right!
I've tried various methods to prepare mellitic acid and while I ran out of lab time before I had finished here are my initial results:
Mellitic acid from Mellite
Mellite is the natural aluminium salt of mellitic acid Al2C12O12.16H2O, F.W. 678.3, it occurs in a few brown coal deposits in Europe and adjacent
parts of Russia. It crystallises as almost octahedral tetragonal bipyramids up to at least 50 mm on edge.
2gm of finely ground natural Mellite from Hungary were dispersed into 10ml of water and 2.5ml of 40% sodium hydroxide added and the suspension boiled
for 20 minutes. Then the solution was just neutralized by adding 5M acetic acid drop-wise from a small pipette and then boiled for a further 20
minutes to precipitate the alumina. The suspension was filtered at the pump resulting in a clear brown solution to which 2.35gm of strontium chloride
dissolved in 10ml of water was added. The precipitate was boiled for a few minutes and allowed to cool.
The creamed coloured precipitate was filter at the pump and washed with several portions of de-ionised water and dried in an oven at 45°C. The result
was a pale cream filter cake of 1.33gm, assuming strontium mellitate is anhydrous this represent 75.6% recovery.

Boffis - 13-7-2011 at 11:50
Strontium mellitate filter cake
The strontium salt was dispersed into 20ml of de-ionised water and 2.3ml of 2M sulphuric acid added. The suspension was boiled for 10 minutes to make
it easier to filter; after this time the heavy granular strontium sulphate precipitate settled rapidly. It was filtered at the pump and the almost
colourless solution slowly evaporated down until an almost dry slurry was obtained, a very small amount of water was added drop-wise until the solid
just dissolved resulting in about 1.5ml of solution. When cool this was placed in a small 5ml beaker and an equal volume of isopropanol added. A white
precipitate formed quickly and the solution was chilled in the fridge for 2 hours and filtered using a very small filter paper disc in a Hirsh funnel.
It was rinsed with isopropanol and sucked almost dry. After oven drying the yield of pure white mellitic acid was 0.13gm or a yield of just 12.9% from
mellite.
There is more acid in the residue but it is very soluble in water and fairly soluble in alcohols. I may be necessary to use a larger portion of
Mellite to make manipulation easier in the final stage.

The final mellitic acid
The next exciting episode will be Ba mellitate from Na diethyl oxaloacetate
UnintentionalChaos - 13-7-2011 at 13:20
Man, this is an old thread of mine...I have learned much in the meantime. How strange to see it being contributed to again. As it turns out, you can
get a decent yield of mellitic acid from hexamethylbenzene (either purchase or if you have a tube furnace... http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv4... ) by oxidation with nitric acid in a sealed tube. I have some hexamethylbenzene
and nitric acid, but no expertise on making/using a sealed tube. Using an open vessel gives very poor yields and I only have a few grams of the
hexamethylbenzene. Time to get reading.
[Edited on 7-13-11 by UnintentionalChaos]
Attachment: C6(Me)6 mellitic via nitric.pdf (363kB)
This file has been downloaded 850 times
Boffis - 13-7-2011 at 20:45
Hi, I have looked at the hexa methylbenzene route and will try it when I can purchase the raw material or have time at home to rig up a tube furnace.
I have the materials and the chemicals, its just time. I fancy trying either dichromate and sulphuric acid or permanganate oxidation as I have lots of
these chemicals and I don't like the idea of heating conc nitric acid in a pressure vessel. The reason I decided to try the route from sodium diethyl
oxaloacetate is simply that I have access to a fare amount of this stuff.
Have you found any data on the mellitic acid salts? I have found data on the acid and natural Al salt (mellite) but data on the salts is hard to find.
I found a bit on the K and NH4 salts in Thorpes dictionary of applied chemistry but very limited data on hydration of salts.
This is where I got to recently before I lost access to my lab for the summer.
Mellitic acid by cyclo-condensation of diethyl oxaloacetate
Theory
Acids and bases catalyse the condensation of carbonyls with an active methylene; that is a –CH2- adjacent to a carbonyl or other electron
withdrawing functional group. Diethyl oxaloacetate contains both functional groups:
Et-O-C:O-C:O-CH2-C:O-O-Et
Therefore it should be capable of condensing with itself; the condensation of three molecules would be capable of cyclotization to produce a
substituted benzene ring. In the case of this ester the most likely cyclical product would be the hexa ethyl ester of mellitic acid though linear
polymerization will be a competing reaction.
Base catalysed condensation is likely to result in rapid hydrolysis of the ester groups too, although this is not a serious issue since the desired
product is free mellitic acid. Acids can also cause such hydrolysis but the process tends to be slower and the resulting ester will generally require
alkali hydrolysis after condensation. Partial hydrolysis is likely to generate acid ester that will act like detergents and tend to cause emulsions
making it difficult to isolate intermediate products.
Diethyl oxaloacetate ester tends to undergo spontaneous condensation over time and form a resin so the compound is general stored as the sodium salt
of the enol tautomer from which it is easily liberated in the cold with dilute acid and the free ester extracted with an organic solvent.
The reaction is as follows:
C8H11O5.Na + HCl = C8H12O5 + NaCl
3C8H12O5 __H+__ C24H30O12 + 3H2O (hydrogen ion is catalytic)
C24H30O12 + 6NaOH = Na6C12O12 + 6C2H5OH
Na6C12O12 + 3BaCl2 = Ba3C12O12 + 6NaCl
Ba3C12O12 + 3H2SO4 = C12H6O12 + 3BaSO4
General Note
Hydration data for the various salt of mellitic acid is hard to come by and it is one of the objects of this work to determine normal hydration of the
more common salts. The strontium and barium salt appear to be either anhydrous or have small hydration numbers or the hydrates are unusually stable.
Experiment 1
4.2 gm (0.02M) of sodium diethyl oxaloacetate were suspended in 15ml of triethylene glycol and 2.2ml of 36% hydrochloric acid (0.025M) added. The
mixture was heated to about 100°C when it began to effervesce freely. The mixture was allowed to reflux until the temperature reached about 150°C
and the mixture had become a homogenous brown liquid. Distil off the volatile components (mostly water) for 30 minutes and then pour into 100ml of
cold water, add 10ml of 40% sodium hydroxide, cool to 10°C and extract with 2 x 30ml portions of ether.
Combine the extracts and distil off the ether. Only a tiny amount of oil brown water dispersible oil remained (<0.2ml), too little to realistically
investigate.
The aqueous phase was evaporated down to about 25ml and acidified with 80% acetic acid. 5gm of strontium chloride hexahydrate dissolved in 15ml of hot
water added and added to the mixture to precipitate any mellitic acid present. Only a trace of precipitate formed, so little that it could not be
removed from the filter paper. The experiment appears to have been a failure.
Note; The triethylene glycol was used as a reaction medium because it always refluxing at a very high temperature however, it appears that the
liberated water controls the reflux temperature. It may have been better to slowly distil the mixture to remove the water and any liberated ethanol
from hydrolysis of the esters. Under these conditions a less volatile acid may be required ie sulphuric acid.
Experiment 2
10.5gm (0.05M) of sodium diethyl oxalylacetate were placed in a 250ml flat bottom flask on a hot plate. 20ml of 30% hydrochloric acid (a little less
than 0.2M) and the mixture refluxed for 2 hours until the amount of brown oil appeared constant and a pale buff precipitate (mainly salt?) settled
rapidly. Cool to room temperature and cautiously add 35ml of 40% (0.35M) of sodium hydroxide (Note 1) and some anti-bumping granules or a few small
porcelain chips (bumping is a problem in the subsequent refluxing) and reflux for a further 2 hours.
Cool to room temperature and dilute with 100ml of water. No oil remained but a small amount of water insoluble solid was removed by filtration. This
material, Product A (Note 2), weighed 0.56gm after washing with water and drying was saved for further investigation.
25gms of barium chloride dehydrate in 150ml of water were added to the clear brown aqueous filtrate (Note 3) and the mixture boiled for a few minutes
to granulate the precipitate. The suspension was filtered at the pump and the precipitated washed with about 30ml of hot water then about 50ml of cold
water and sucked as dry as possible. The pale buff coloured cake was dried at 45°C for 24 hours. The yield of barium salt was 7.06 gm of pale buff
hard brittle filter cake (product B). If this is the barium salt of mellitic acid and the barium salt is anhydrous this would represent a yield of
56%.
TO BE CONTINUED When I get access to my lab again in September
Note 1 because some of the ester may have undergone partial hydrolysis as mentioned above no attempt was made to isolate the intermediate compound.
Note 2 a small portion of Product A was heated in small spoon, it melted and eventually took fire and burned with a smoky flame leaving a little sooty
black residue. The material is clearly organic but time did not permit an investigation into this material. It may be a random polymerization product
or un-hydrolyzed hexa-ethyl mellitate.
Note 3 it may be better to neutralise the filtrate with acetic acid to reduce co-precipitation of barium carbonate and hydroxide. Strontium chloride
is preferable to barium chloride because when the corresponding salt is decomposed with dilute sulphuric acid strontium sulphate has a much smaller
tendency to occlude organic molecules than barium sulphate and the yield may be significantly reduced as a result.
UnintentionalChaos - 13-7-2011 at 21:05
As noted in the paper I attached, KMnO4 is very prone to destroying the product. I seriously doubt the quoted quantitative yield of mellitic acid by
room temp KMnO4 if only 30-60C under the same conditions destroyed most of the product.
In synthesizing phthalic acid from o-xylene with hot KMnO4, I also had significant destruction of product, which does not bode well for the related
mellitic surviving. Nitric acid is unique here in that it is quite poor at oxidizing past the acid.
Boffis - 13-7-2011 at 21:26
I still think KMnO4 might be worth a try for the patient! I am often away for a couple of months at a time so I might try it just before I go away!
Dichromate /H2SO4 has similar problems.
I must admit I have used nitric acid oxidation of Inisitol to rhodizonic acid and it is a surprisingly clean oxidizing agent
Now all I need is some hexa methylbenzene!
[Edited on 14-7-2011 by Boffis]