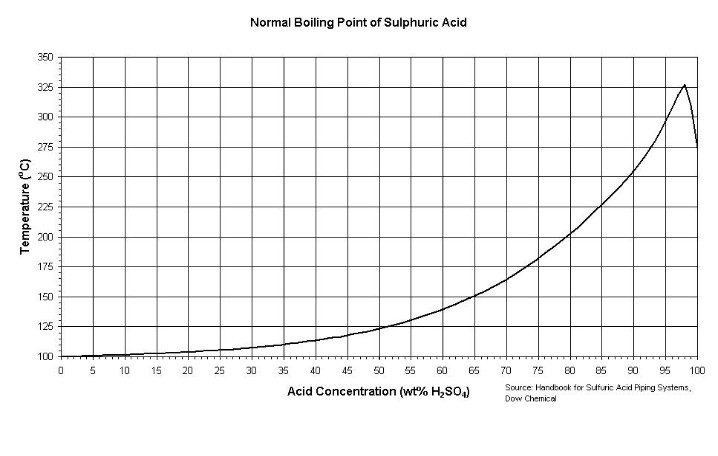
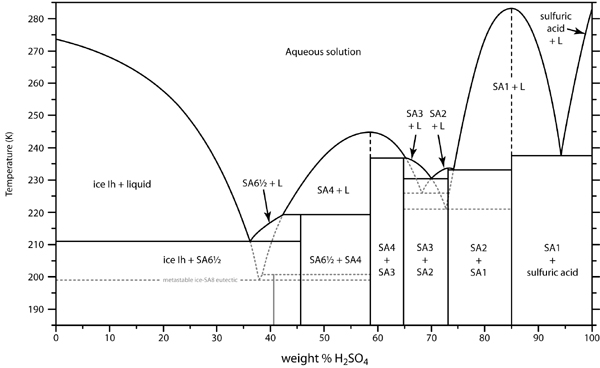
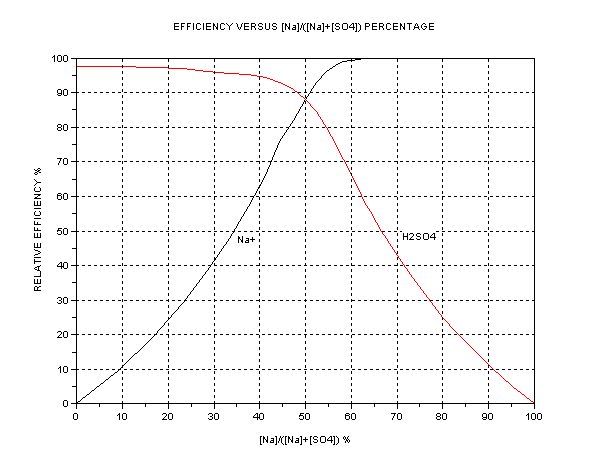
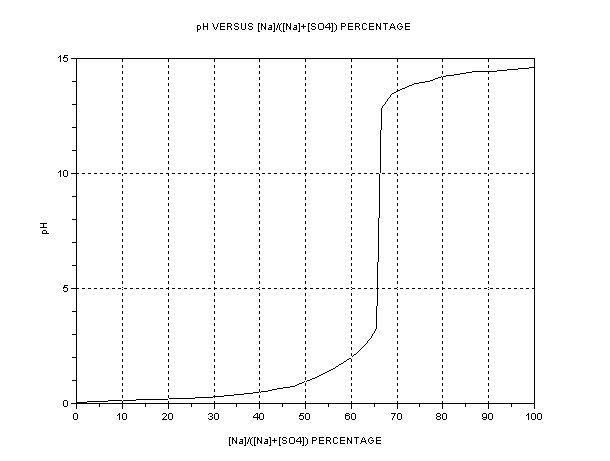
| In the anode cell, then, the relative mobility (directly related to conductivity) of the H+ ions is about 350 and HSO4- about 50, (units 10^-4 m^2
mho/mol).(CRC). For every HSO4- ion passing through the barrier, 7 H+ ions go the other way, per 8 electrons delivered to the anode. This means that
7 HSO4- ions deliver their electron from the anode solution for every one that comes from the cathode area. |