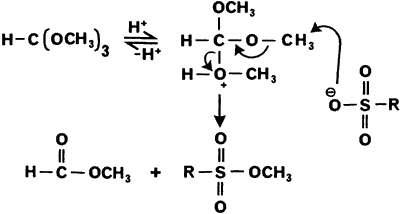
250 g of toluenesulfonic acid and 50 g of sulfur are suspended in carbon tetrachloride and treated with stirring with chlorine. The resultant heat
development is prevented by external cooling. Sulfur gradually goes into solution under simultaneous escape of hydrogen chloride and sulfur dioxide.
After completion of chlorine absorption water is added, partly in order to decompose excess sulfur chloride, and partly in order to remove dissolved
hydrochloric acid and sulfurous acid. The carbon tetrachloride solution leaves behind after the evaporation of the solvent, 250 g of nearly pure
sulfonic acid chloride, which corresponds to a yield of 90 per cent. Judging from pure p-toluenesulfonic acid, we obtain the p-toluenesulfonyl
chloride, mp 68°.
|