Originally posted by PainKilla
Ritter, I think you are ignoring literature precedents yourself. The paper that describes the toxic intermediates of bromomethylation (did you read it?) clearly states that such intermediates do not persist through
the reaction. On top of that, haloethers hydrolyze very rapidly upon a slight rise in pH, so the workup will very quickly eliminate any sort of
dangerous intermediates.
By the way, do you really consider reactions that involve carbene intermediates safe?
On top of that, the point is not to synthesize 4-hydroxybenzaldehyde using literature precedents but to discuss potential methods of its synthesis. It
would be even cheaper to buy 4-hydroxybenzaldehyde, but I see no one has suggested that (yet).
As far as the bromomethylation is concerned... I don't think bromomethylation is quite selective enough to yield only para-substituted product....
Although, the tosyl group may be big enough to promote mostly para product. I think this is one of those things that only experiment can say for sure.
I found a nice paper that seems to indicate that benzyl bromides may be convered into their aldehydes by refluxing in ethanol-H2O2-KBr (attached).
------
http://www.google.com/patents?id=5nZwAAAAEBAJ&printsec=a...
I'll see if I can find any references to phenol (or its relatives) halomethylation.
----(no access)----
Haloalkylation of aromatic compounds. III. Chloro- and bromomethylation of phenolic ethers.
Tourinho, Ana M.; Da Rocha, Nilmar V. P.
Anais da Associacao Brasileira de Quimica (1967) 26(3-4):19-22.
Journal written in Portuguese.
Abstract:
The chloromethyl derivs. of PhOR (where R = Me, Et, Pr, Bu, or C5H11) were prepd. in 48-70% yields by their reaction at room temp. with HCHO and HCl
in AcOH, H2SO4 being slowly added. The bromomethyl derivs. were obtained by corresponding methods in 52.5-60% yields.
--------
Chloromethylation of alkoxybenzenes and the preparation of the corresponding benzaldehydes. Very Relevant!
Journal fuer Praktische Chemie (Leipzig) (1956) 3:274-7.
Abstract
The following p-alkoxybenzyl chlorides, p-ROC6H4CH2Cl (I), were prepd. at 15-20 by addn. of 1 mole ROPh in C6H6 to 48 g. paraformaldehyde in 200 ml.
concd. HCl (R, % yield, and b.p./mm. given): Et, 71,120-4/12; Pr, 75, 134/12; iso-Pr, 63, 126-8/14; Bu, 65, 138-42/10; iso-Bu, 54, 98/0.8; Am, 59,
154-8/12; and iso-Am, 64, 146-8/12. Oxidation of I to the corresponding ROC6H4CO2H by refluxing with 50 g. HNO3 (35 B.acte.e.) 10 hrs. served for the
identification of the a products. I with AcOH and hexamethylenetetramine (cf. Sommelet, C.A. 8, 660) produced the p-ROC6H4CHO: Et, 61, 119-22/10; Pr,
72, 129-30/10; iso-Pr, 69, 122-3/10; Bu, 70, 140-1/10; iso-Bu, 66, 152/16; Am, 61, 156-8/10; and iso-Am, 67, 147-8/10. Refluxing 0.5 mole I in 800
ml. Me2CO with 4.5 g. NaI and 33 g. NaCN gave the 4-ROC6H4CH2CN (II): Et, 84, 150-1/11; Pr, 79, 162/12; iso-Pr, 67, 154/14; Bu, 69, 174-8/15; iso-Bu,
64, 158-9/12; Am, 74, 176-8/10; iso-Am, 74, 194/22. Refluxing 0.2 mole II in 100 ml. EtOH, 32 ml. water, and 32 g. KOH gave the 4-ROC6H4CH2CO2H (R, %
yield, and m.p.): Et, 78, 91.3; Pr, 63, 91; iso-Pr, 83, 87; Bu, 77, 88.5; iso-Bu, 82, 87; Am, 79, 86; and iso-Am, 88, 87.
(71% yield of the benzyl bromide from ethoxybenzene... probably means even better can be obtained with bromomethylation w/ R-OTs).
------
From what I saw, chloromethylation procedures carried out at room temperature, gave good yields of the para-product from variously substituted phenol
ethers (methoxy, ethoxy etc). I think with -OTs, a carefully carried out bromomethylation will work quite well, given this information. Phenol itself
is too reactive for the direct halomethylation (see patent).
Also, can someone please get the Chloromethylation of alkoxybenzenes and the preparation of the corresponding benzaldehydes
reference? I would be very interested in seeing it, as I am sure would anyone else interested in this thread/halomethylation.
[Edited on 22-6-2008 by PainKilla] |

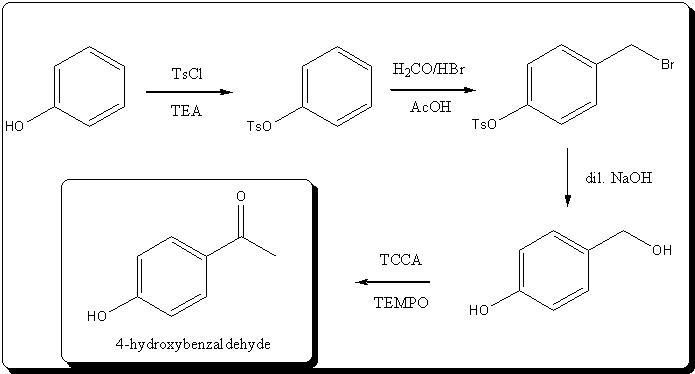
 ) to the aldehyde.
) to the aldehyde.
 for a total yield of 56.5%...
for a total yield of 56.5%...