Chelating agents like EDTA are quite non-discriminating: they form strong complexes with many (most, in fact) non-alkali metals, including Fe and Nd.
U and Nd being in the same column is really a meaningless coincidence. Nd belongs to the First f-block (Lanthanides), which show very strong
inter-group chemical similarities. U belongs to the Second f-block (Actinides), which show also strong inter-group chemical similarities (but less so
than Lanthanides).
Due to their electronic configuration structures, both groups also show similarities between each other.
Similarity between U and Nd however is really quite weak. Yes, both form EDTA complexes but then so do... erm... Al and Pb! U forms oxidation states
from +2 to +6 (but +3 isn't very stable), Nd only does +3. U forms very stable complex anions and cations, Nd only forms complexed Nd(+3) cations.
By contrast, the Fe/Nd separation methods presented and tested to destruction in this thread are simple to use and require mostly only OTC chemicals.
That's why we chose them...
[Edited on 11-6-2015 by blogfast25] |








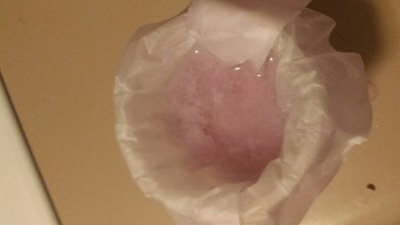

 ). Based on earlier discussions in this thread, I processed a half-inch cube Nd magnet using the oxalate separation method: demagnetize, remove
plating, dissolve in sulfuric acid, filter, oxidize all Fe(II) to Fe(III), precipitate Nd2(Ox)3 with oxalic acid. That worked fairly well, I think,
as it has for other people in this thread. Later, I was planning on converting the oxalate to the oxide, then redissolving in H2SO4 for the final
sulfate.
). Based on earlier discussions in this thread, I processed a half-inch cube Nd magnet using the oxalate separation method: demagnetize, remove
plating, dissolve in sulfuric acid, filter, oxidize all Fe(II) to Fe(III), precipitate Nd2(Ox)3 with oxalic acid. That worked fairly well, I think,
as it has for other people in this thread. Later, I was planning on converting the oxalate to the oxide, then redissolving in H2SO4 for the final
sulfate.